DCS-120 Confocal and Multiphoton FLIM Systems
Abstract: The DCS-120 system uses excitation by ps
diode lasers, femtosecond fibre lasers or femtosecond titanium-sapphire lasers,
fast scanning by galvanometer mirrors, confocal detection, and FLIM by bhs multidimensional
TCSPC technique to record fluorescence lifetime images at high temporal
resolution, high spatial resolution, and high sensitivity [1, 2]. The DCS‑120 system is available with inverted microscopes
of Nikon, Zeiss, and Olympus. It can also be used to convert an existing
conventional microscope into a fully functional confocal or multiphoton laser
scanning microscope with TCSPC detection. Due to its fast beam scanning and its
high sensitivity the DCS-120 system is compatible with live-cell imaging. The
DCS-120 covers the whole range from basic FLIM recording to advanced
multi-dimensional FLIM applications. Advanced applications include simultaneous
recording of FLIM or steady-state fluorescence images simultaneously in two
fully parallel wavelength channels, laser wavelength multiplexing, time-series
recording, Z stack FLIM, phosphorescence lifetime imaging (PLIM), fluorescence
lifetime-transient scanning (FLITS) and FCS recording. Applications focus on
lifetime variations by interactions of fluorophores with their molecular
environment. Typical applications are ion concentration measurement, FRET
experiments, metabolic imaging, and plant physiology.
DCS-120 Versions for any Kind of Application
The DCS-120 systems are complete laser
scanning microscopes for fluorescence lifetime imaging. The systems use bhs
multi-dimensional TCSPC FLIM technology [1, 3, 7, 14, 17] in combination with
fast laser scanning and confocal detection or multi-photon excitation [8]. DCS-120 systems are available with
various inverted and upright microscopes, see Fig. 1 and Fig. 2. Moreover, the
DCS-120 scan head with the associated control and data acquisition electronics
can be used to upgrade a conventional microscope with scanning and FLIM recording.
In the basic configuration, the DCS-120 uses excitation by two ps diode lasers
and records in two parallel detector and TCSPC channels. Advanced versions of
the DCS-120 system are available with multiphoton excitation by Ti:Sa lasers
and femtosecond fibre lasers (Fig. 2, bottom). The system also works with
tuneable excitation sources[29, 30]. A DCS-120 MACRO system [1, 9] is
available for FLIM of centimetre-size objects, see Fig. 2, second row, right.

Fig. 1: The DCS‑120 system with a Zeiss Axio Observer microscope

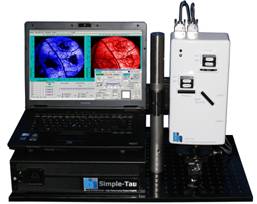


Fig. 2: Upper
row: DCS-120 Axio Observer system, DCS‑120 MACRO system. Lower row:
DCS-120 MP multiphoton system with Ti:Sa laser, DCS-120 MP
multiphoton with femtosecond fibre laser
The DCS systems are using highly efficient
GaAsP hybrid detectors, combining extremely high efficiency with large active
area, high counting speed, short acquisition time, high time-resolution, and
low background [1]. The DCS-120 system is using 64-bit data acquisition
software [31], resulting in FLIM at unprecedented pixel numbers. All system
components, including lasers, scanner, microscope, and detectors are controlled
by one piece of software, making the system easy to use. FLIM data analysis is
performed by bh's legendary SPCImage software [5, 4]. It combines time-domain
and phasor analysis, uses an MLE algorithm to fit the data, and runs the calculation
on a GPU, resulting in data processing times of no more than a few seconds.
These features make the DCS-120 system superior to other systems even in
entry-level FLIM applications.
However, this is not all. The bh FLIM
technique is based on a new understanding of FLIM in general [1]. FLIM is not
just considered a way to add additional contrast to microscopy images. Instead,
it is considered and designed as a molecular imaging technique. bh FLIM
exploits the fact that the fluorescence decay function of a fluorophore is an
indicator of its molecular environment, and that multi-exponential decay
analysis delivers molecular information, such as the metabolic state of live
cells and tissues, protein conformation and protein interaction, reaction of
cells to drugs and molecular environment, or mechanisms of cancer development
and cancer progression. To reach this target, bh FLIM systems have features not
available by other systems: Compatibility with live-cell imaging,
extraordinarily high time resolution and photon efficiency, capability to split
decay functions into several components, excitation-wavelength multiplexing in
combination with parallel-channel detection, recording of dynamic lifetime
effects caused by fast physiological effects, and simultaneous FLIM/PLIM [1].
Principle of Data
Acquisition
Multi-Dimensional TCSPC
The bh FLIM systems use a combination of
bhs multidimensional time-correlated single-photon counting process with
confocal or multiphoton laser scanning. The sample is continuously scanned by a
high-repetition rate pulsed laser beam. Single photons of the fluorescence
signal are detected, and each photon is characterised by its time in the laser
pulse period and the coordinates of the laser spot in the scanning area in the
moment of its detection. The recording process builds up a photon distribution
over these parameters, see Fig. 3. The photon distribution can be interpreted
as an array of pixels, each containing a full fluorescence decay curve in a
large number of time channels. In the DCS system, two such recording channels
are used in parallel to record images in different spectral intervals or under
different polarisation.
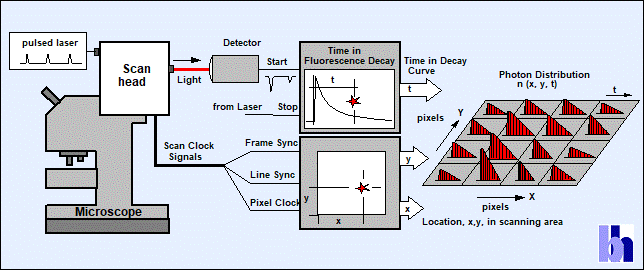
Fig. 3:
Principle of TCSPC FLIM
The recording process delivers a near-ideal
photon efficiency, excellent time resolution, and is independent of the speed
of the scanner. The signal-to-noise ratio depends only on the total acquisition
time and the photon rate available from the sample.
The technique can be extended by including
additional parameters in the photon distribution. These can be the depth of the
focus in the sample, the wavelength of the photons, the time after a
stimulation of the sample, or the time within the period of an additional modulation
of the laser. These techniques are used to record Z stacks or mosaics of FLIM
images, multi-wavelength FLIM images, images of physiological effects occurring
in the sample, or to record simultaneously fluorescence and phosphorescence
lifetime images.
Optical Principle
One-Photon Scanning
The traditional way of laser scanning
microscopy is excitation via the traditional one-photon process. That means
individual fluorophore molecules are excited by absorbing just one photon of
the excitation light at a time. The absorption / emission process is a linear
one, i.e. doubled excitation power also induces double emission from the
fluorophores. As a result, one-photon excitation excites light in a double cone
through the entire depth of the sample, see Fig. 1, left. This causes the
commonly known 'out-of-focus blur' in conventional microscopy. To obtain a
clean image from a defined focal plane suppression of out-of-focus light is
required. The traditional way to solve the problem is laser scanning with
'Confocal Detection'.
Confocal scanning is based on deflecting
the excitation beam by fast moving galvanometer mirrors, focusing the beam into
the sample via the microscope lens, and feeding the fluorescence from the
sample back through the microscope lens and the scan mirrors through a pinhole
in a plane conjugate with the focal plane in the sample. Only light from the
focal plane passes the pinhole. An image built up by a detector behind the
pinhole is free of out-of-focus light and laterally scattered light. Confocal
scanning thus delivers extremely clean images of a defined image plane inside
an object. This is important especially for FLIM because, once recorded, decay
components from unwanted sample planes are hard to remove from the data. The
principle of the scanner is shown in Fig. 4.
Two laser beams of different wavelength are
coupled into the scanner. They are combined by a beam combiner, pass the main
beamsplitter, and are deflected by the scan mirrors. The scan lens sends the beam down the microscope beam path in a way
that the scan mirror axis is projected into the back aperture of the microscope
lens. The motion of the scan mirrors causes a variable tilt of the beam in the
plane of the microscope lens. The laser is thus scanning an image area in the
focal plane of the microscope lens. The scanning can be very fast - the line
time can be as short as a millisecond, an entire frame can be scanned in less
than a second.
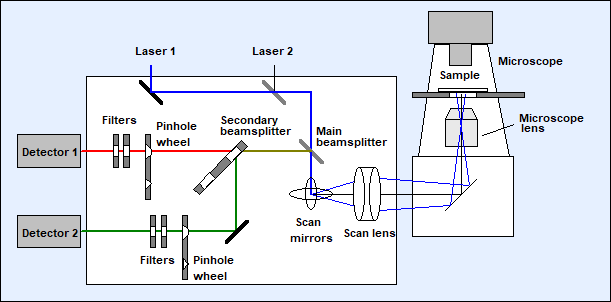
Fig. 4:
Optical diagram of the DCS-120 scan head. Simplified, see [2] for details
The fluorescence light is collected back
through the microscope lens, passes the scan lens, and is again reflected at
the scan mirrors. The reflected beam is stationary, independently of the motion
of the scan mirrors. It is separated into two spectral or polarisation
components, and projected into confocal pinholes. The light signals passing the
pinholes are filtered spectrally, and sent to the detectors. Only light from
the excited spot in the focal plane of the microscope lens reaches the
detectors. The result is a clear image from a defined depth inside the sample
which is free of out-of-focus blur and lateral scattering.
MACRO Scanning
The DCS-120 scan head can be used with a
wide variety of inverted and upright microscopes of different manufacturers,
provided these have an optical port that makes the upper image plane of the microscope
lens available. The DCS-120 scan head can, however, also be used without a
microscope. In this version of the system, the 'DCS-120 MACRO', the sample is
placed directly in the image plane of the scan lens [1, 9]. The system then scans
objects as large as 20 mm in diameter.
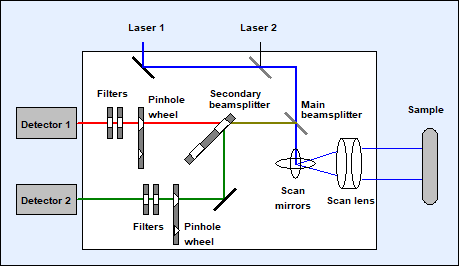
Fig. 5: DCS-120 MACRO system. It scans a
sample directly in the image plane of the scan lens. Samples as large as 20 mm
can be scanned.
The problem of one-photon excitation is
that visible or UV wavelengths have to be used for excitation. The absorption
at these wavelengths is high, so that the efficiency decreases rapidly with
increasing focus depth in the sample. One-photon scanning therefore cannot be
used to image deep layers in biological tissue, see Fig. 6, left. The solution
to tissue imaging is multiphoton scanning by a titanium-sapphire laser or a
femtosecond fibre laser. Different than confocal scanning, which avoids out-of
focus detection, multiphoton scanning avoids out-of-focus excitation. By
exciting the fluorophore molecules by a multiphoton (usually two-photon)
process only molecules in the focus of the laser are excited, see Fig. 6,
middle. Therefore, no pinhole is needed to restrict detection to a defined
image plane. The fluorescence light can be fed directly, without passing back
through the scanner, to the detectors. This makes it possible to detect
fluorescence light which is scattered on the way out of the sample (Fig. 6,
right). Moreover, the laser wavelength is in the NIR, where absorption and
scattering coefficients are lower than in the visible or UV range.
Consequently, deep layers of the sample can be reached. The capability to
excite in and detect from deep sample layers makes multiphoton scanning the
choice for tissue imaging. Another advantage of multiphoton excitation is that fluorophores
with absorption in the UV can be reached without the need of UV optics.
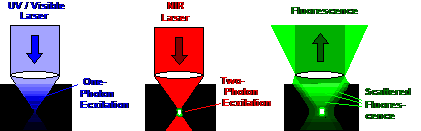
Fig. 6: Comparison of one-photon
excitation and multiphoton excitation. Left: The one-photon process excites
within a full double cone throughout the sample. The effective excitation power
decreases rapidly with increasing depth. Middle: Two-photon excitation excites
only in the focus of the laser beam. The NIR laser penetrates deeply into the
sample. Right: The fluorescence from a deep focus is scattered on the way out
of the sample. It leaves the back aperture of the microscope lens in a wide
cone. It cannot be detected via a confocal beam path
but very well via NDD.
The principle of
the DCS system in the multiphoton configuration is shown in Fig. 7. The beam of
a Ti:Sa laser or of a femtosecond fibre laser is fed into the scanner through
one of the two laser ports. It can be combined with a visible laser connected
to the other port, but this is not a condition for multiphoton operation. The
laser beam is deflected by the galvanometer mirrors, and focused in the sample
by the scan lens and the microscope lens. The fluorescence light is collected
though the microscope lens. However, it is not sent back through the scanner.
Instead, it is diverted from the microscope beam path by a dichroic mirror,
filtered and/or split into spectral components by a secondary beamsplitter, and
fed to one or two detectors. The principle is called 'Non-Descanned Detection'.
The FLIM data are built up from the photon pulses of these detectors as
described in Fig. 3.
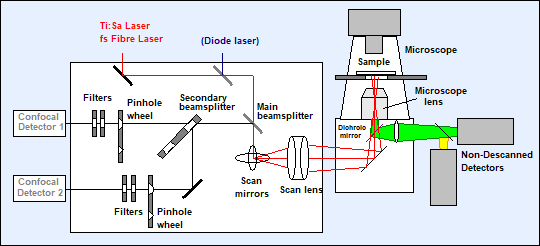
Fig. 7: Scanning with 2-photon excitation. Non-descanned detectors shown
on the right.
On-photon
excitation, two-photon excitation and descanned and non-descanned detectors can
be combined in one DCS-120 system. In that case, a ps diode laser is injected
via the second laser port, and the one-photon images are detected by confocal
detectors. By enabling either the non-descanned detectors or the confocal
detectors the system can be switched from one-photon and multiphoton operation
and vice versa.
TCSPC Modules
Different
DCS-120 versions can contain different TCSPC / FLIM modules. Early DCS systems
used SPC-150 modules. From 2016 on SPC150 N and SPC-150 NX modules
were used. The SPC-150 N, and, especially, the SPC-150 NX achieve
higher time resolution in combination with ultra-fast hybrid detectors and
femtosecond lasers. Recent systems use either the SPC‑180 NX or the
SPC-QC-104. The SPC-180 NX delivers maximum time resolution with fast
detectors and lasers, the SPC-QC has lower time resolution but can record at
extremely high count rates. Still, the instrument response (IRF) of the SPC‑QC 104
is faster than the pulse width of diode lasers and faster than the transit-time
spread of most detectors, see Fig. 9. Hence there is little difference in
resolution for confocal systems with diode lasers. For systems with femtosecond
lasers, however, it can be the difference between easily detecting a fast decay
component and missing it.


Fig. 8: SPC-180 NX (left) and
SPC-QC-104 (right)
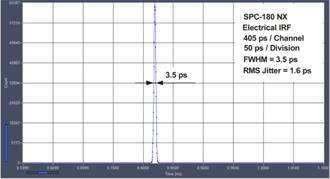
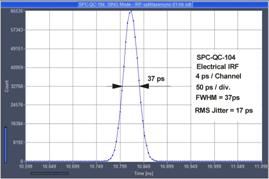
Fig. 9: Electrical IRF for SPC-180 NX (left) and SPC-QC 104
General Features of the
DCS-120 System
Precision Confocal and Multiphoton FLIM Images
By using confocal or multiphoton laser
scanning and multi-dimensional TCSPC, the DCS system combines the two most
precise techniques of recording in space and in time. FLIM images recorded by
the DCS-120 systems feature diffraction-limited spatial resolution, ultra-high
temporal resolution, suppression of out-of focus fluorescence, suppression of
longitudinal and lateral scattering, optical sectioning capability, near-ideal
sensitivity and photon efficiency, and low background. By recoding FLIM images
with extraordinarily high pixel numbers and time-channel numbers the results
are free of undersampling artefacts.
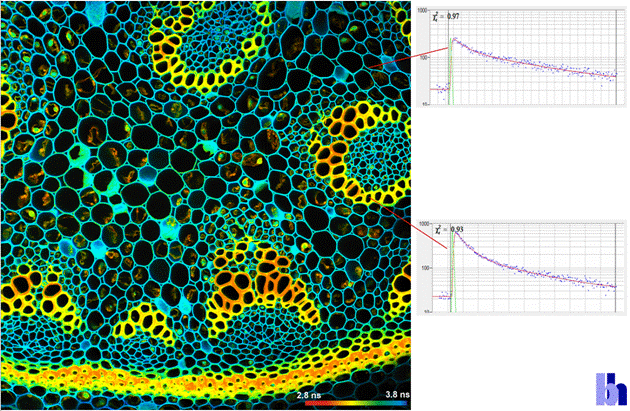
Fig. 10: Megapixel FLIM image recorded by the
DCS-120 system. Decay curves in selected pixels on the right.
Ultra-High Efficiency
The bh HPM‑100‑40 GaAsP hybrid
detectors of the DCS‑120 combine ultra-high sensitivity with the large
active area of a PMT [27]. The
large area avoids any alignment problems, and allows light to be efficiently
collected through large pinholes and from the non-descanned beam path of the
DCS-120 MP system [2]. In contrast to conventional PMTs or SPADs there is no
secondary peak or diffusion tail in the temporal response. Importantly, the
hybrid detectors are free of afterpulsing. The absence of afterpulsing results
in improved contrast, higher dynamic range of the decay curves recorded, and in
the capability to obtain FCS data from a single detector. The combination of
these features makes it easy to detect fluorescence from endogenous
fluorophores in single cells and split the decay curves into several decay
components, see Fig. 11.
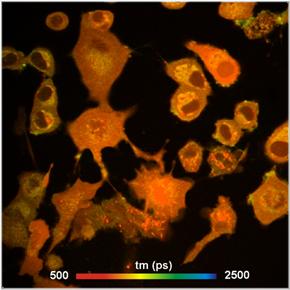
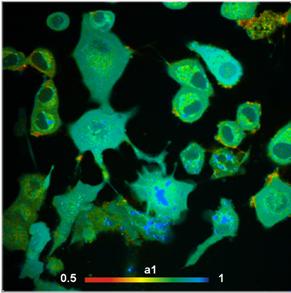
Fig. 11: Autofluorescence lifetime image of NADH in live cells. Lifetime
image of mean lifetime of double exponential decay (left) and image of
amplitude of fast decay component, a1 (metabolic indicator, right).
The Ultimate in Time Resolution and Timing Stability
The electrical time resolution of the
SPC-180 NX FLIM modules is 3.5ps fwhm, or about 1.5 ps rms [1]. Timing stability is better than 0.4ps
rms. The system IRF of a multiphoton system with an HPM-100-06 detector is
<19 ps fwhm, or 8.3 ps rms, including detector and laser. Please
see Fig. 12. A FLIM example is shown in Fig. 13. The
fast decay component, t1, has a lifetime of 10 ps.
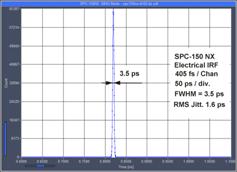

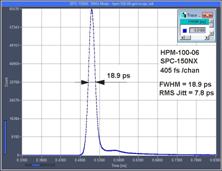
Fig. 12: Electrical IRF, IRF stability, and system IRF with ultra-fast
detectors and femtosecond laser excitation

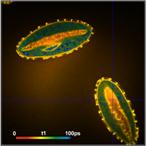
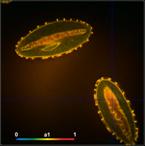
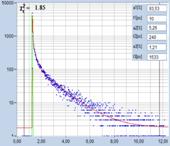
Fig. 13: FLIM of Pollen grains with a dominating decay component of 10 ps.
DCS-120 MP, fibre laser, HPM-100-06 fast hybrid detector
Wide Range of Excitation Wavelengths
The DCS-120 confocal system can be used
with a wide range of excitation wavelengths. Available diode-laser wavelengths
range from 375 nm for excitation of NADH to 785 nm for excitation of
NIR dyes. An NADH (autofluorescence) image is shown in Fig. 14, an image of a
pig skin sample incubated with 3,3-diethylthiatricarbocyanine in Fig. 15.
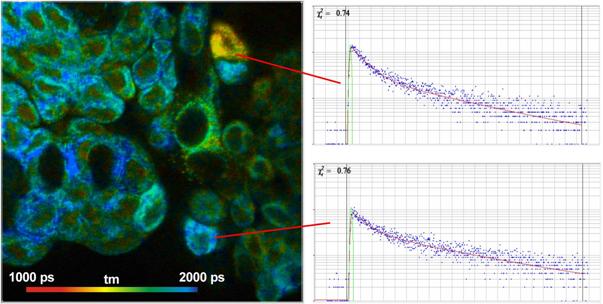
Fig. 14:
UV-Excitation FLIM. NADH image of cells, excitation 370 nm, detection 420
to 475 nm.
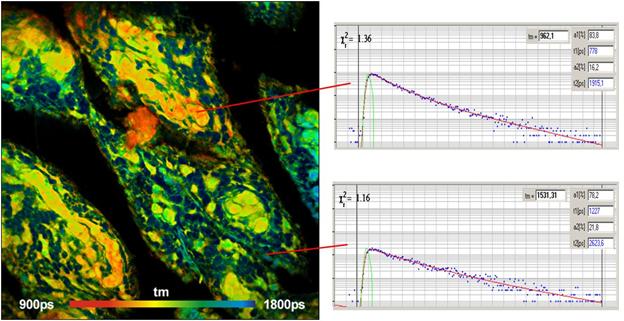
Fig. 15: Near-Infrared FLIM. Pig skin sample stained with
3,3-diethylthiatricarbocyanine, detection wavelength, excitation 690 nm,
detection wavelength from 780 nm to 900 nm.
Fast Beam Scanning - Fast Acquisition
The DCS-120 uses fast beam scanning by galvanometer
mirrors. A complete frame is scanned within a time from 100 ms to a few
seconds, with pixel dwell times down to one microsecond.
Compared with sample scanning, beam
scanning is not only much faster, it avoids also induction of cell motion by
exerting dynamic forces on the sample. Moreover, live cell imaging requires a
fast preview function for sample positioning and focusing. This can only be provided
if the beam is scanned at a high frame rate.
With its fast scanner and its
multi-dimensional TCSPC process the DCS system achieves surprisingly short acquisition
times. An autofluorescence FLIM image of a live Enchytraeus albidus is
shown in Fig. 16. The acquisition time was 1 second. Considering the large
pixel number and the high signal-to-noise ratio this is faster than what is
achieved by many 'Fast FLIM' techniques [1, 45].
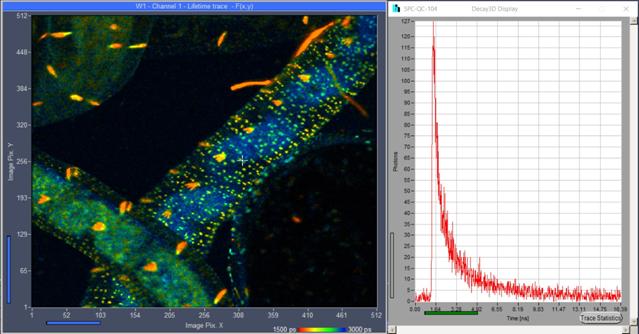
Fig. 16: Lifetime image taken from a live Enchytraeus albidus.
Autofluorescence, 1 second acquisition time at 8 MHz average count rate and 80 MHz
laser repetition rate. DCS-120 system with SPC‑QC‑104. Online FLIM
with SPCM software. Decay curve in selected pixel shown on the right.
Fast scanning also improves the options for
time-series recording. The change in the fluorescence lifetime of the
chloroplasts in a moss leaf with the time of exposure Fig. 17. Time per image
is 1 second.

Fig. 17: Change of the fluorescence lifetime of chlorophyll with time of
exposure. Moss leaf, excitation at 445 nm, 256 x 256 pixels, 1
image per second.
Megapixel FLIM Images in
Two Parallel Channels
With 64 bit SPCM software pixel numbers can
be increased to 2048 x 2048 pixels, with a temporal resolution of 256
time channels. The DCS-120 system is able to simultaneously record two
high-resolution images in different wavelength or polarisation channels, see Fig.
18 and Fig. 19.
Fig. 18: BPAE sample (Invitrogen)
scanned with 2048 x 2048 pixels. Green channel, 485 to 560 nm
Recording is performed in two fully
parallel TCSPC channels, avoiding any electronic lifetime or intensity
crosstalk. Even if one channel should saturate the other is still producing
correct data.
The capability to record images of large
pixel numbers is beneficial for a wide range of FLIM applications. One example
is tissue imaging where the samples are large, and the images are containing a
wealth of detail. It is also useful when a large number of cells have to be
investigated and the FLIM results be compared. Megapixel FLIM records images of
many cells simultaneously, and under exactly identical environment conditions.
Moreover, the data are analysed in a single analysis run, with identical IRFs
and fit parameters. The results are therefore exactly comparable for all cells
in the image area.
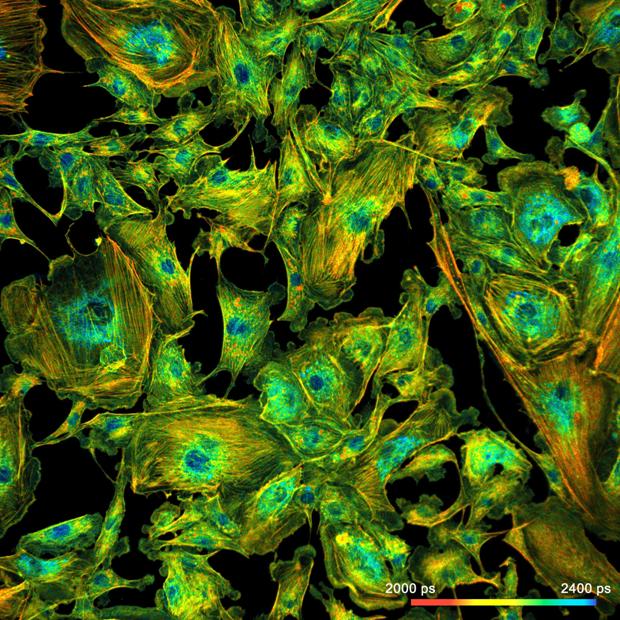
Fig. 19: BPAE sample (Invitrogen),
scanned with 2048 x 2048 pixels. Red channel, 560 to 650 nm
High Image Contrast up to the Highest Count Rates
Images taken with conventional TCSPC FLIM
at high count rate often suffer from low intensity contrast. The reason is not
the pile-up effect, as commonly believed, but intensity nonlinearity by the
dead time of the TCSPC process. DCS-120 systems using the SPC-180 or SPC-QC-104
modules do not show this effect. The SPC-180 takes the intensity information
from a fast parallel counter with almost no dead time, the SPC-QC-104 avoids
dead time by a fast time-conversion method. An image taken at an average count
rate of 5.5 MHz is shown in Fig. 20. Peak count rate is about 10 MHz. No
loss in contrast by dead time effects is visible.
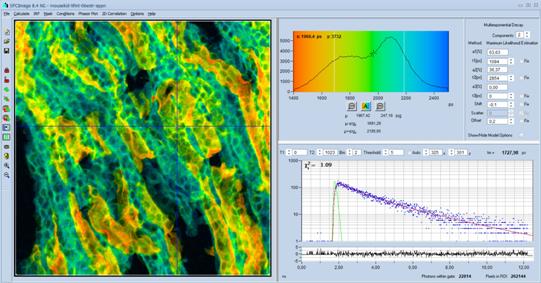
Fig. 20: Image taken at an average count rate of 5.5 MHz. Peak count
rate is about 10 MHz. No loss in contrast by dead time effects is visible.
DCS-120 with SPC-180 NX module, data analysis by SPCImage NG.
Photon-Counting Intensity Images
A frequently asked question is whether the
DCS system can record conventional intensity images. Of course it can - the number
of photons, and thus the intensity is part of the FLIM information contained in
every pixel of a lifetime image. Recent DCS systems using SPC-180 or SPC-QC-104
TCSPC modules even deliver the intensity without dead-time-induced nonlinearity.
Intensity images can be displayed side by side with lifetime images, see Fig. 21.
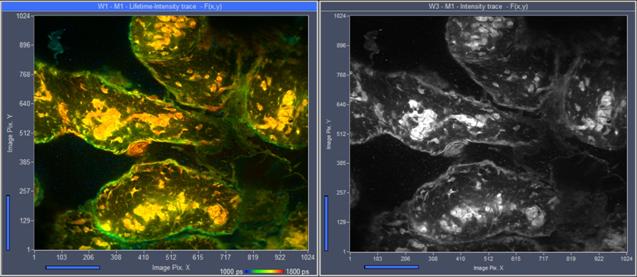
Fig. 21: Lifetime image (left) and intensity image (right), simultaneously
displayed by SPCM. SPC-180N, lifetime-intensity mode.
Online Display of Lifetime Images and Decay Curves
The SPCM software is able to display
lifetime images and decay curves online during the measurement. Online display
of lifetime data helps the user evaluate the quality of the data recorded and,
if necessary, restart the measurement in a different region of the sample, with
different zoom of the scanner, or with different pinhole size or laser power.
An example of online display is shown in Fig. 22.
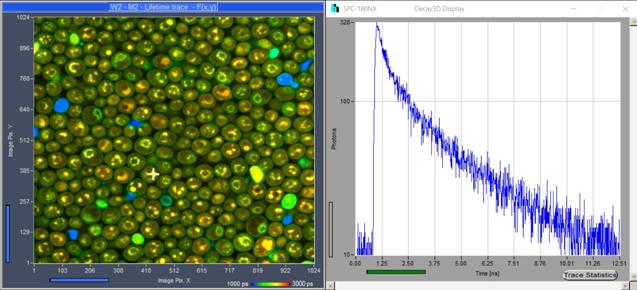
Fig. 22: Online display of lifetime image and decay curve.
Multiphoton FLIM with Ti:Sa Laser
The DCS-120 system is available with
multiphoton excitation. The beam of a Titanium-Sapphire laser is fed into one
of the laser ports of the DCS scan head. Laser power control and on/off
modulation is achieved via an acousto-optical modulator (AOM). Laser control is
embedded in the DCS data acquisition software, see 'Software', page 45. FLIM
images of pig skin in different depth of the tissue are shown in Fig. 23.
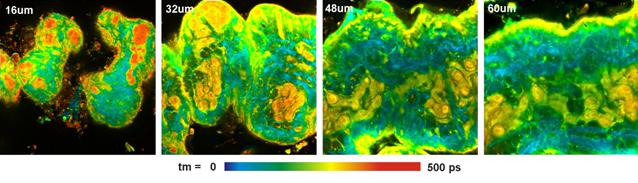
Fig. 23: Pig skin, NADH autofluorescence, image in different depth in the
sample. Amplitude-weighted lifetime of triple-exponential decay model
Multiphoton FLIM with Fibre Laser
The DCS-120 can be combined with a
femtosecond fibre laser [11, 12]. The preferred wavelength is 780 nm,
making the DCS-120 Fibre system perfectly suitable for NADH imaging in tissue.
For other fluorophores, the lack of tuneability may be considered a
disadvantage. It turns out, however, that most of the commonly used exogenous
fluorophores can be excited at reasonable efficiency. This makes the DCS-120
the cheapest multiphoton laser scanning microscope on the market.
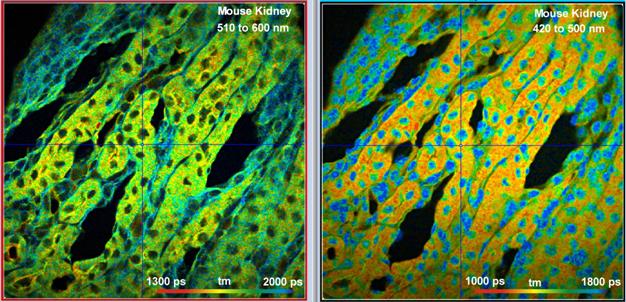
Fig. 24: Mouse kidney sample labelled with Alexa 488, Alexa 568, and DAPI.
DCS-120 Fibre, Images simultaneously detected in two spectral channels. Image
format 1024 x 1024 pixels, 1024 time channels. Excitation power 3 mW in the
sample plane, count rate about 2×106 s-1 in each
channel.
Non-Descanned Detection
The advantage of multiphoton excitation is
that it penetrates deeply into biological tissue. Multiphoton FLIM is therefore
the method of choice for molecular imaging in deep tissue. However,
fluorescence from deep layers is scattered on its way out of the sample and
does not pass back through the scanner. Therefore its is diverted from the
excitation beam path before it re-enters the scanner, and sent to
'Non-Descanned' detectors, please see Fig. 25. Scattered photons from the
excited spot are detected by the NDD detectors, and assigned to the current
pixel by the TCSPC imaging process. The result is high efficiency for image
planes located deeply inside tissue. An example is shown in Fig. 26.

Fig. 25: Principle of non-descanned detection
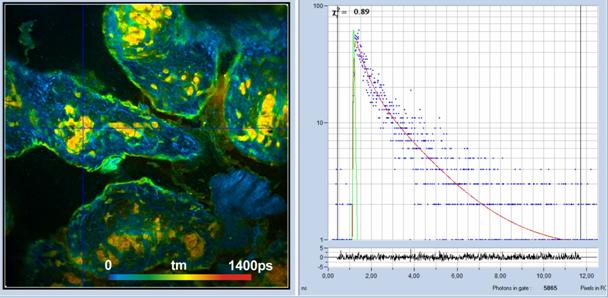
Fig. 26: FLIM of pig skin, NADH image, DCS-120 MP Fibre system, two-photon
excitation, non-descanned detection.
Express FLIM
Recording a typical TCSPC image within a
short period of time requires an enormous data transfer rate from the TCSPC
module to the computer. The data transfer problem increases if a longer
sequence of images is to be recorded, and if several TCSPC channels are
operated at high count rate simultaneously. bh have solved the problem by a
technique called 'Express FLIM'. Express FLIM does not transfer data into the
computer photon by photon. Instead, the hardware of the TCSPC module combines
the information of all photons within a given pixel into a just two numbers.
One is the first moment of the decay curve, the other the number of photons
within the pixel. Both numbers are transferred to the computer at the end of
each pixel. Even for fast scanning, the required data transfer rate can easily
be achieved. The result is a lifetime image that contains first-moment values
in the individual pixels. Express FLIM is available for all DCS systems
containing the SPC-QC-104 module. An example is shown in Fig. 27.
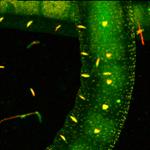
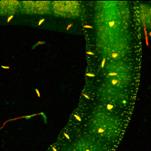
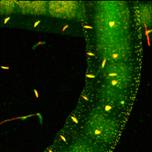
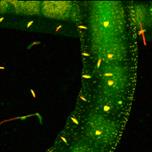
Fig. 27: Express-FLIM of a live Enchytraeus albidus. Autofluorescence, four
subsequent images from a 5-frames/second sequence. DCS-120 system with
SPC-QC-104. Excitation pulse rate 80 MHz, average photon rate about 10×106 s-1.
FLIM of Macroscopic Objects
The DCS-120 MACRO system records
lifetime images of objects as large as 20 mm in diameter. The system does not
use a microscope. Instead, the object is placed directly in the primary image
plane of the scanner. For autofluorescence applications the DCS MACRO scan head
is available with a scan lens of especially high UV transmission. An example of
a MACRO FLIM image is shown in Fig. 28.
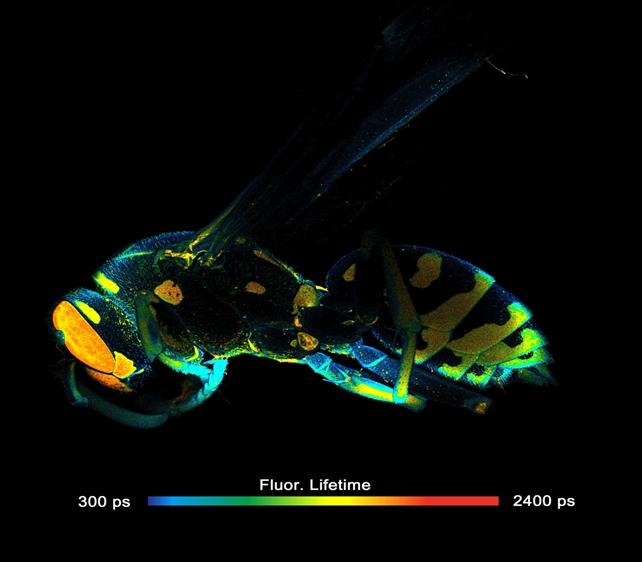
Fig. 28: High-resolution MACRO FLIM image. 2048 x 2048 pixels,
256 time channels per pixel.
FCS
With its
time-tag, or, more precisely, parameter-tag mode the DCS-120 confocal system
delivers highly efficient FCS. Because the hybrid detectors are free of
afterpulsing there is no afterpulsing peak in autocorrelation data [27]. It is
not necessary to suppress the afterpulsing peak by cross-correlation, resulting
in an increase of the signal-to-noise ratio [1, 16]. An example of FCS is shown
in Fig. 29.
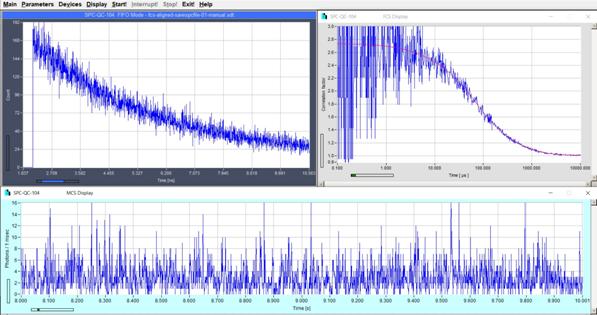
Fig. 29: Single molecules diffusing through the laser focus. Decay curve,
FCS curve, intensity trace. Raman light suppressed by time-gating. Online fit with FCS procedures of SPCM data acquisition software.
Detection of Nanoparticles
The parameter-tag mode of the DCS system
can also be used for single-particle detection. An example for the diffusion of
fluorescent nanoparticles through the laser focus in shown in Fig. 30.
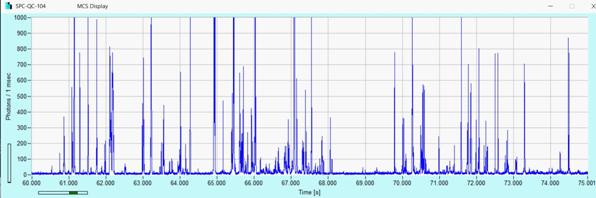
Fig. 30:
Fluorescent nanoparticles drifting through the laser focus. Intensity trace.
The individual photon bursts can be further
analysed by bh 'SPCDynamics' software, see Fig. 31. SPCDynamics displays
fluorescence decay curves integrated over the bursts in a selected time
interval, a phasor plot of the decay data of the bursts within a selected time
interval, and decay curves and fluorescence lifetimes of individually selected
bursts.
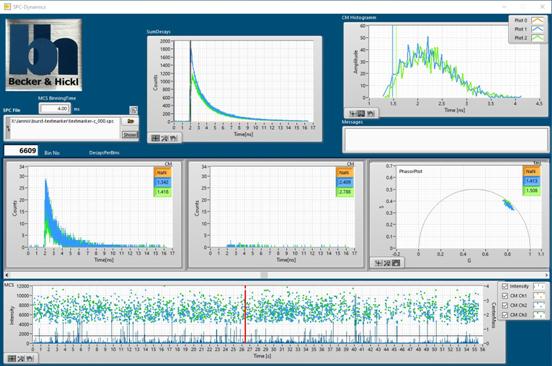
Fig. 31:
Analysing photon bursts from single particles or single molecules by bh
'SPCDynamics' software
Recording of Single Decay Curves
The DCS-120 system can record single decay
curves from fluorophore solutions or from selected spots in a two-dimensional
sample. Curves are obtained either in the 'Single' mode of SPCM, or from
summing up the decay data from a pixel area within a FLIM image. This makes a
separate lifetime spectrometer for fluorophore characterisation unnecessary. Moreover:
A multiphoton system with ultra-fast detectors beats any lifetime spectrometer
in time resolution. An example is shown in Fig. 32.
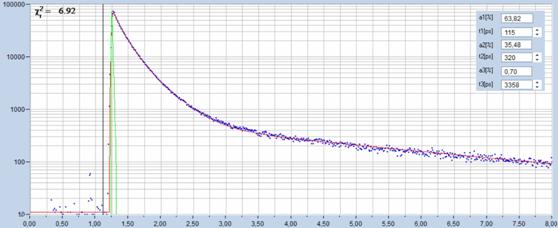
Fig. 32: Fluorescence decay curve recorded with a DCS-120 MP.
Advanced DCS‑120
Functions
Mosaic FLIM
Mosaic FLIM records a large number of
consecutive images into a single FLIM data array. The individual images within
this array can represent the elements of a tile scan (x-y mosaic), images in
different depth in the sample (z-stack mosaic), or images for different times
after a stimulation of the sample (temporal mosaic). An example of an x-y
mosaic is shown in Fig. 33. The complete data array has 2048 x 2048
pixels, and 256 time channels per pixel. Compared to a similar image taken
through a low-magnification lens the advantage of mosaic FLIM is that a lens of
high numerical aperture can be used, resulting in high detection efficiency and
high spatial resolution.
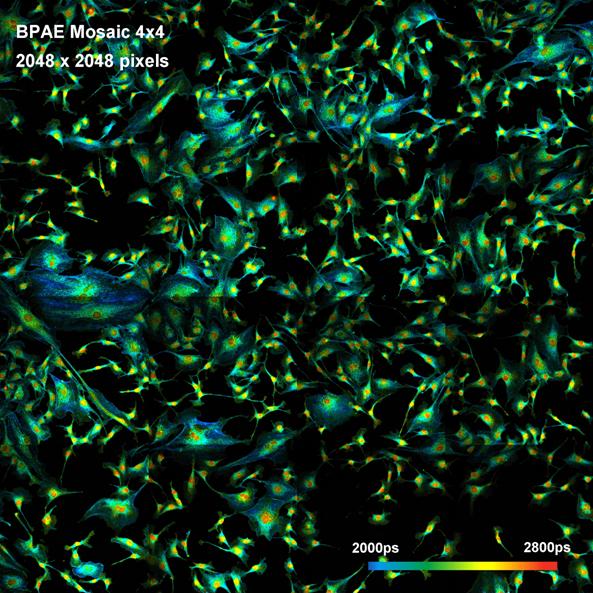
Fig. 33: Mosaic FLIM of a BPAE sample. The mosaic has 4x4 elements, each
element has 512x512 pixels with 256 time channels. The entire mosaic has
2048 x 2048 pixels, each pixel holding 256 time channels. DCS‑120
MP multiphoton system with motorised sample stage.
Temporal Mosaic FLIM: Recording of Dynamic Effects
SPCM 64-bit software versions later than
2014 have a Mosaic Imaging function implemented. For time-series recording,
subsequent frames of the scan are recorded into subsequent elements of the mosaic.
The sequence can be repeated and accumulated [1, 8]. The time per mosaic
element can be as short as a single frame, which can be less than 100 ms.
Another advantage is that the entire array can be analysed in a single SPCImage
data analysis run. Fig. 34 shows the change of the lifetime of chlorophyll in
plant tissue with the time of illumination.
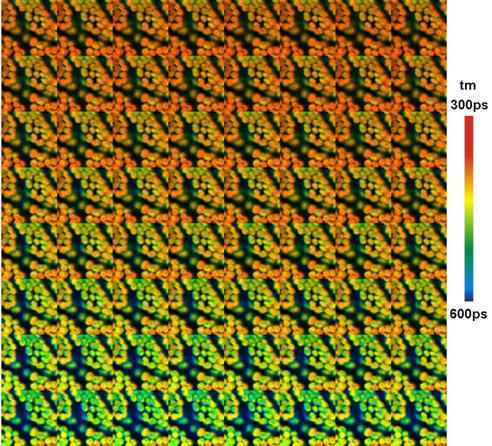
Fig. 34: Time series of chloroplasts in a leaf recorded by Mosaic Imaging.
64 mosaic elements, each 128x128 pixels, 256 time channels. Scan time per
element 1s. Experiment time from lower left to upper right. Amplitude-weighted
lifetime of double-exponential decay.
Temporal Mosaic FLIM with Triggered Accumulation
When dynamic effects in the fluorescence
behaviour of an object are to be recorded the speed is limited by the decrease
of the photon numbers in the pixels. The DCS system solves the problem by
'Triggered Accumulation'. A dynamic effect in the measurement object is induced
periodically, and the start of the mosaic recording is synchronised with the
stimulation. The photon number in the pixels then only depends on the total
acquisition time (the number of stimulation periods), and not on the speed of
the mosaic recording. As a result, a very fast image sequence can be obtained
without the need of exceedingly high photon rate. In fact, triggered
accumulation FLIM can be faster than any 'fast FLIM' technique and still be
live-cell friendly. An example is shown below.
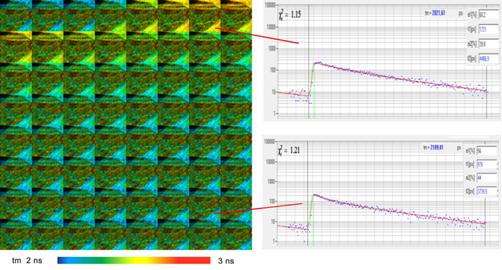
Fig. 35, Left:
Calcium transient in cultured neurons, temporal mosaic imaging, 40 ms per
image. Image elements 64x64 pixels. bh SPC-150 FLIM system with SPCM software,
attached to a Zeiss LSM 7MP microscope.
FLIM of Moving Objects
The recording of fluorescence-lifetime
images of live cells or organisms is often impaired by motion in the sample.
Nevertheless, the DCS system is able to obtain precision fluorescence-lifetime data from such
objects. The technique is based on temporal-mosaic recording and image
segmentation by the phasor plot of the bh SPCImage NG data analysis software. A
cluster of phasors is selected in the phasor space, identifying pixels of a
given decay signature in the FLIM mosaic. These pixels are back-annotated in
the mosaic, selecting parts of the objects irrespectively of their location in
the individual images. The decay data of the pixels within the selected areas
are summed up. The result is a single decay curve with extremely high pixel
number which can be analysed at high precision [5, 4].
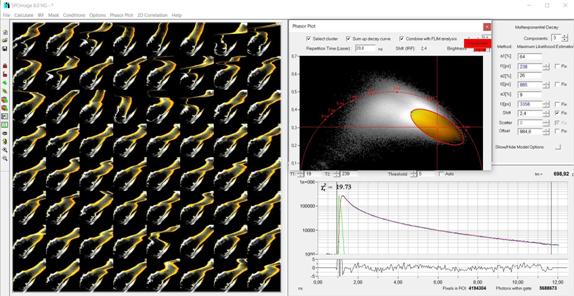
Fig. 36: Precision lifetime analysis
on a moving object. A water flee is imaged by temporal mosaic FLIM (left), the
phasor range of a structure of interest is selected, and fluorescence-decay
analysis is performed on the decay data of the combined pixels within the
phasor range.
FLITS: Fluorescence Lifetime-Transient Scanning
FLITS records dynamic effects in the fluorescence
lifetime of a sample along a one-dimensional scan. The technique is based on
building up a photon distribution over the distance along the scan, the arrival
times of the photons after the excitation pulses, and the experiment time after
a stimulation of the sample. The maximum resolution at which lifetime changes
can be recorded is given by the line scan time. With repetitive stimulation and
triggered accumulation transient lifetime effects can be resolved at a
resolution of about one millisecond [1, 18, 28,].
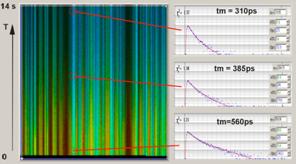
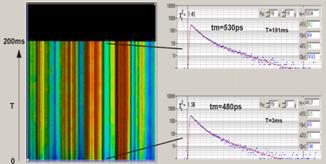
Fig. 37: FLITS of chloroplasts in a
grass blade, change of fluorescence lifetime after start of illumination. Left:
Non-photochemical transient, transient resolution 60 ms. Right:
Photochemical transient. Triggered accumulation, transient resolution
1 ms.
Z Stack recording
Z stack recording is achieved by
controlling the Z drive of the microscope, usually a Zeiss Axio Observer or
Axio Examiner, synchronously with the acquisition of an image sequence. The DCS
system has two procedures to record Z stacks. One is based on Mosaic FLIM: The
data of subsequent planes are recorded in a large FLIM Mosaic. An example of a
Mosaic-FLIM Z stack is shown in Fig. 38.
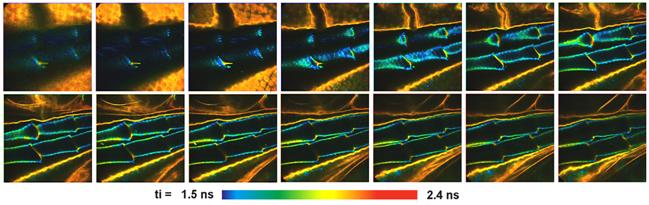
Fig. 38: FLIM Z stack of a part of a water flee. Z stack by mosaic FLIM
procedure.
The advantage of Mosaic Z stack recording
is that the images of all Z planes are recorded in a single, large FLIM data
set. This avoids delay by writing data in subsequent data sets, and guarantees
that the data of all planes are exactly comparable. However, the maximum number
of planes is limited by the available data space. Depending on the desired
lateral resolution, that means that 16 to 64 Z planes can be recorded.
A virtually unlimited number of Z planes
can be recorded by a 'record and save' procedure. That means an image of the
current plane is recorded, saved into a file, and then the focal plane is moved
to the next Z plane. The procedure is repeated until the desired number of
planes has been recorded. The record-and-save procedure needs time to save the
data but is able to record large number of planes at high x-y resolution. An
example of a high-resolution Z stack obtained from a fly, Musca domestica,
is shown below. The stack contains 289 planes, each scanned with 1024 x1024
pixels and 1024 time channels. Fig. 39 shows a projection of all planes in a
single FLIM image by the 'Multi-File View' of SPCM.
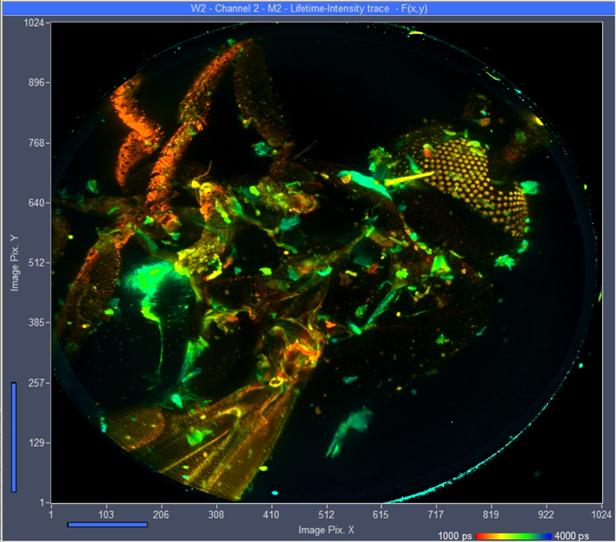
Fig. 39:
Vertical projection of all 289 planes of the z stack into a single FLIM image.
Planes added by Multi-File View of SPCM, image displayed by
Online-Lifetime function of SPCM. Single-exponential lifetime by first-moment
analysis.
Fig. 40 shows the same data, analysed by
SPCImage and combined into a 3D representation by Image J. All 289 planes
were processed with a double-exponential model by the batch-processing function
of SPCImage NG, and the resulting 289 tm images written into bmp files by the
batch-export function. The bmp files were imported into Image J, which then
constructed the 3D representation. For details please see [10].
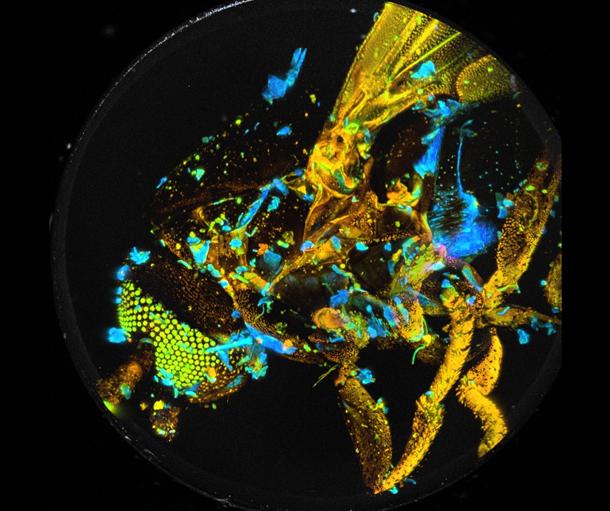
Fig. 40: Decay analysis by SPCImage NG and 3D reconstruction by ImageJ.
Colour represents mean lifetime, tm, of double-exponential decay, lifetime
range red to blue = 0 to 1250 ps.
Multi-Wavelength FLIM
With the bh multispectral FLIM detectors
the DCS‑120 records FLIM simultaneously in 16 wavelength channels [1, 3, 7, 13, 15]. The images are recorded by a
multi-dimensional TCSPC process which uses the wavelength of the photons as a
coordinate of the photon distribution. There is no time gating, no wavelength
scanning and, consequently, no loss of photons in this process. The system thus
reaches near-ideal recording efficiency. Moreover, dynamic effects in the
sample or photobleaching do not cause distortions in the spectra or decay
functions. Multi-wavelength FLIM got an additional push from the new 64-bit
SPCM software, and from the introduction of a highly efficient GaAsP
multi-wavelength detector [1]. 64-bit software works with enormously large
photon distributions, and the GaAsP detector delivers the efficiency to fill
them with photons. As a result, 16 images in 16 wavelength channels can be
recorded at a resolution of 512x512 pixels and 256 time channels. An example is
shown in Fig. 41. For applications please see [1, 37, 47].
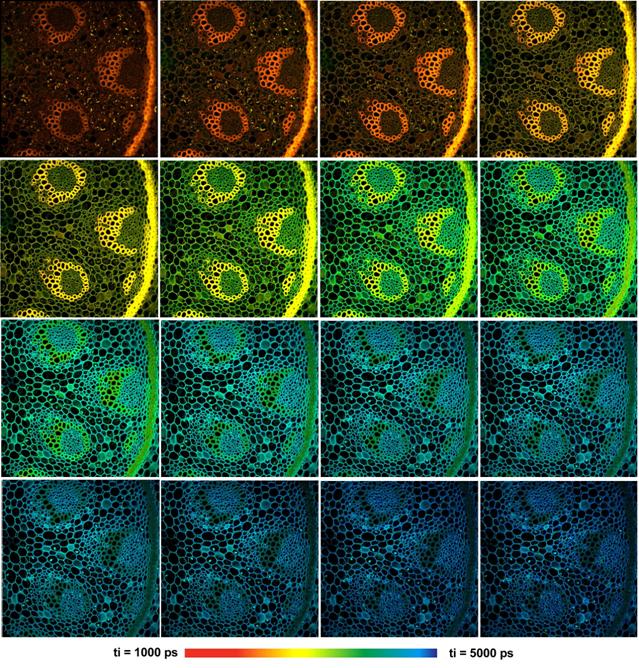
Fig. 41: Multi-wavelength FLIM with the
bh MW-FLIM GaAsP 16-channel detector. 16 images with 512 x 512 pixels
and 256 time channels were recorded simultaneously. Wavelength from upper left
to lower right, 490 nm to 690 nm, 12.5 nm per image. DCS‑120
confocal scanner, Zeiss Axio Observer microscope, x20 NA=0.5 air lens.
It may be suspected that the spatial and temporal
resolution of the individual images is mediocre, at best. This is, however, not
the case, thanks to the large data space available in the 64-bit environment.
Fig. 42 demonstrates the true spatial resolution of the data. Images from two
wavelength channels, 502 nm and 565 nm, were selected from the data
shown Fig. 41, and displayed at larger scale and with individually adjusted
lifetime ranges. With 512x512 pixels and 256 time channels, the spatial and
temporal resolution of the individual images is comparable with what previously
could be reached for FLIM at a single wavelength. Decay curves for selected
pixels of the images are shown in Fig. 43.
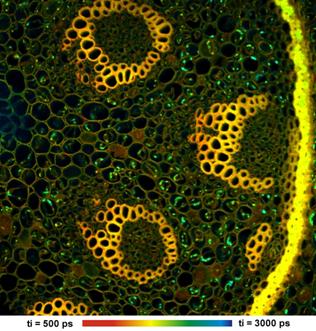

Fig. 42: Two images from the array shown in Fig. 41, displayed in larger
scale and with individually adjusted lifetime range. Wavelength channels
502 nm (left) and 565 nm (right). The images have 512 x 512
pixels and 256 time channels.
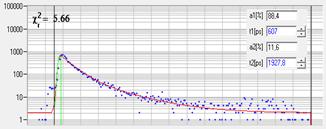
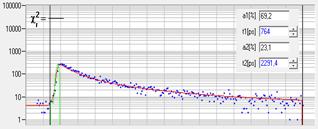
Fig. 43: Decay curves at selected pixel position in the images shown above.
Blue dots: Photon numbers in the time channels. Red curve: Fit with a
double-exponential model.
Laser multiplexing is used to record FLIM
images from fluorophores that cannot be excited at the same wavelength, or the
fluorescence of which cannot be discriminated by emission filters. Two, three,
or four lasers can be multiplexed in time. Multiplexing is automatically
synchronised with the pixels, lines or frames of the scan. The data acquisition
software builds up separate images for the individual lasers. An example is
shown in Fig. 44. It shows a mouse kidney section, stained with Alexa 488 WGA,
Alexa 568 phalloidin, and DAPI. Two excitation wavelengths, 405 nm and
473 nm, were multiplexed. The detection wavelength intervals were
432 nm to 510 nm and 510 nm to 550 nm. Only the
combinations of 405 nm with 432 nm
to 510 nm and 510 nm to 550 nm, and 473 nm and
510 nm to 550 nm are shown. The fourth combination, 473 nm with
432 nm to 510 nm does not contain reasonable data because the
detection interval is too close to the excitation wavelength.
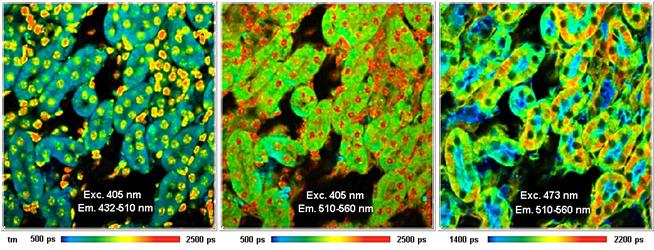
Fig. 44: Excitation wavelength multiplexing, 405 nm and 473 nm.
Detection wavelength 432 nm to 510 nm and 510 nm to 550 nm.
Mouse kidney section, stained with Alexa 488 WGA, Alexa 568 phalloidin, and
DAPI.
The DCS-120 records FLIM and PLIM by bh's
simultaneous FLIM/PLIM technique. The technique is based on on/off modulation
of the excitation laser, and recording of lifetime images for the photon times
in the laser pulse period and in the laser modulation period [1, 32]. On/off
modulation is defined by the laser control parameters. The recording process is
synchronised with the modulation via the GVD-120 or -140 Scan controller.
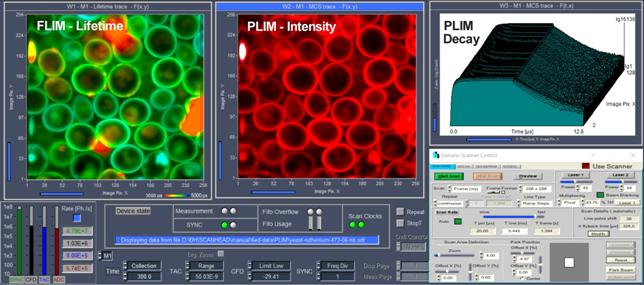
Fig. 45: Simultaneous FLIM / PLIM,
main panel of data acquisition software
Applications in Life
Sciences
Molecular Imaging
FLIM uses the fluorescence decay function
of a fluorophore as an indicator of its molecular environment. The fluorescence
decay function, within reasonable limits, neither depends on the fluorophore
concentration nor on the excitation power, or other instrumental details. This
is a striking advantage over intensity-based imaging techniques. When
fluorescence in a sample is excited (Fig. 46, left) the emission intensity
(second left) depends not only on possible interaction of the fluorophore with
the molecular environment but also on the fluorophore concentration, on
possible absorption in the sample, on the excitation power, and on the light
collection efficiency of the optics. Changes in the molecular environment can
thus not be distinguished from changes in these parameters. Spectral
measurements (second right) are able to distinguish between different
fluorophores. However, changes in the local environment usually do not cause
changes in the shape of the spectrum. The fluorescence lifetime of a
fluorophore however (Fig. 46, right), only depends on the fluorophore itself
and its interaction the molecular environment.
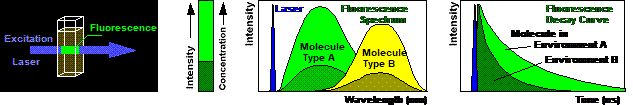
Fig. 46: Fluorescence. Left to right: Excitation light is absorbed by a
fluorophore, and fluorescence is emitted at a longer wavelength. The
fluorescence intensity varies with concentration. The fluorescence spectrum is
characteristic of the type of the fluorophore. The fluorescence decay function
is an indicator of interaction of the fluorophore with its molecular
environment.
By using the fluorescence lifetime, or,
more precisely, the shape of the fluorescence decay function, molecular effects
can therefore be investigated independently of the unknown and usually variable
fluorophore concentration [1, 44].
Frequent FLIM applications are ion
concentration measurements, probing of protein interaction via FRET, and the
probing of the metabolic state and the cell viability via the fluorescence
decay parameters of NADH and FAD.
Fluorescence decay functions in these applications are usually multi-exponential. The components
of the decay function represent different binding states, different
conformations of the fluorophore, or other biologically relevant information. Highly
efficient multi-exponential fluorescence-decay analysis is therefore an
integral part of the DCS-120 system [4, 5, 6].
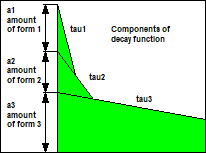
Fig. 47: Real decay function of a
fluorophore in biological environment (left) and composition of the curve
(right).
Molecular Parameters - Derived from Fluorescence-Decay
Data
Molecular environment parameters, such as
local pH, ion concentrations, local viscosity or redox potential are available
through TCSPC FLIM and precision decay analysis [1]. With appropriate
calibration of the probe the results are quantitative, i.e. independent of the
laser power, the fluorophore concentration, the parameters of the
optical-system, and other instrumental details. Examples are shown below.
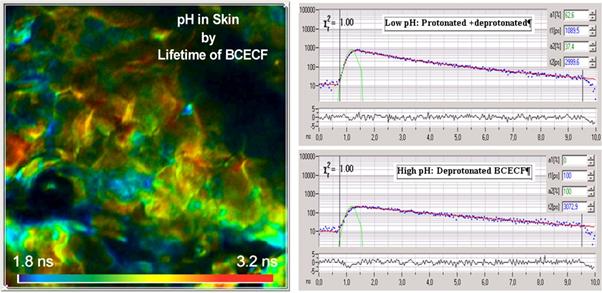
Fig.
48: pH in skin, detected by lifetime of BCECF
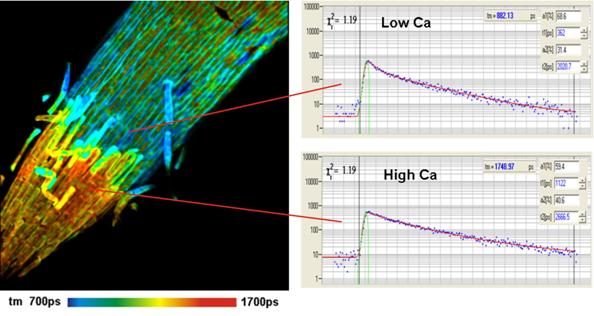
Fig. 49: Calcium concentration in barley
root. Detected by lifetime of Oregon Green Bapta
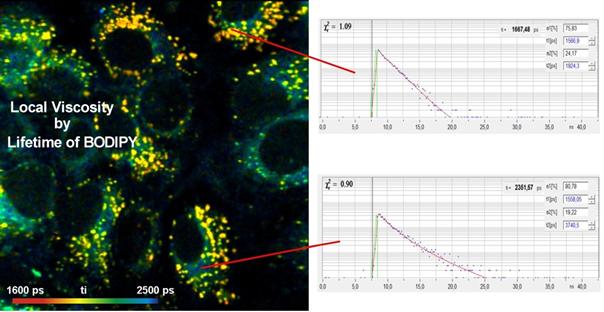
Fig. 50: Local viscosity, detected by
lifetime of BODIPY
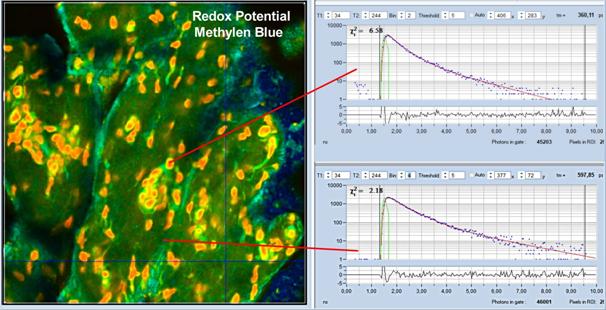
Fig. 51: Redox potential, detected by
lifetime of Methylen Blue
FRET - Results from a
Single Donor FLIM Image
FRET (Förster Resonance Energy Transfer) is used to probe protein
interaction and protein structure in biological systems. The excitation energy
is absorbed by a donor. It then transfers to an acceptor, and is emitted via
the emission band of the acceptor. The energy transfer occurs only if the
distance between the donor and the acceptor is less than a few nm. FRET is used
to obtain information about protein interaction, protein folding, and protein
structure. FRET experiments by steady-state spectroscopy are difficult to
calibrate. FRET is therefore performed mainly by FLIM. But also with FLIM there
are problems if the measurement is based on simple single-exponential
'fluorescence lifetimes'. Single-exponential FRET is often considered a
quantitative technique but in fact it is not [21].
Quantitative FRET results are only obtained by FLIM in combination
with double-exponential FRET analysis. The method has been developed by bh in 2005
and has been constantly improved in the past years. In contrast to
single-exponential techniques, the method delivers correct FRET efficiencies
and FRET distances even for incomplete donor-acceptor linking, and without
reference measurement of a donor-only sample [1, 22]. The classic FRET efficiency, the FRET
efficiency of the interacting donor, the amount of interacting donor, and the
donor-acceptor distance are displayed directly by SPCImage NG [4]. An example
is shown in Fig. 52.
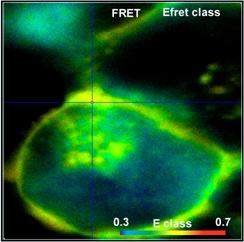
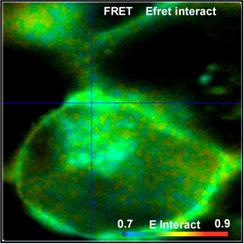
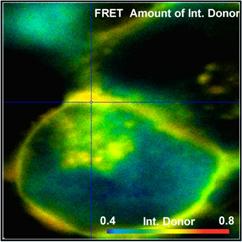
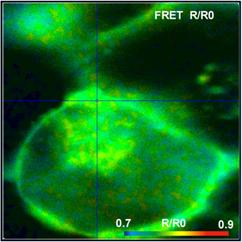
Fig. 52: FRET Measurement in life cell. Classic FRET efficiency, FRET
efficiency of interacting donor, amount of interacting donor, and ratio of
donor-acceptor distance to Förster radius. bh double-exponential FRET
technique, DCS-120 confocal, SPCImage NG data analysis.
FRET-Based Sensors
A large group of sensors for cell
parameters is based on FRET. A donor and an acceptor are attached to the ends
of a amino-acid linker. The linker changes its conformation with the molecular
environment, and so does the fluorescence decay of the donor. For a summary
please see FRET chapter in [1]. Fig. 53 shows an open tumor in a mouse,
expressing a sensor for apoptosis. The sensor consist of mKate2 (donor) and
iRFP (acceptor), connected by an amino-acid linker [48].
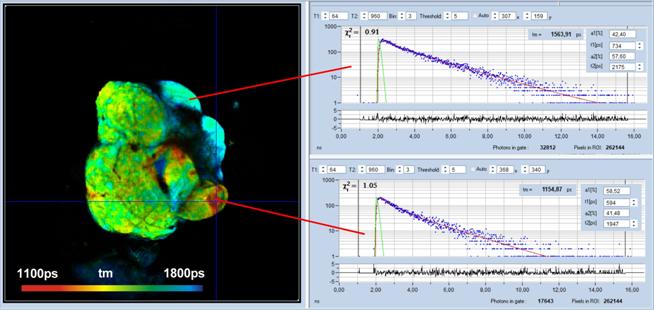
Fig. 53: Open tumor in a mouse, expressing a FRET sensor for apoptosis.
DCS-120 MACRO system, analysis by SPCImage NG
Label-Free Imaging
FLIM of Small Organisms
The wide range of excitation and detection
wavelengths and the high sensitivity makes the DCS-120 an excellent system for label-free
(autofluorescence) FLIM of small organisms. Fig. 54 shows an autofluorescence
image of Artemia salinas, a small shrimp living in briny water. A
two-photon FLIM image of Artemia salinas recorded by the DCS‑120 MP
Fibre system [36] is shown in Fig. 55.
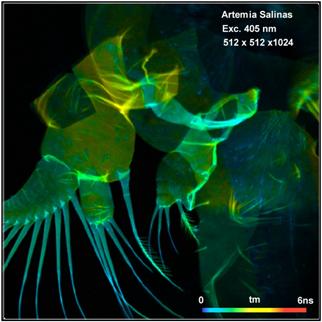
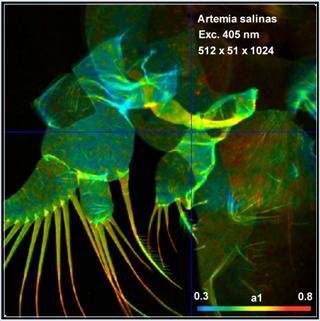
Fig. 54: Autofluorescence FLIM of Artemia salinas. Left:
Amplitude-weighted lifetime, tm. Right: Metabolic parameter, a1. DCS-120
confocal system with HPM‑100‑40 hybrid detectors and SPC-180 TCSPC
modules, Analysis by SPCImage NG.
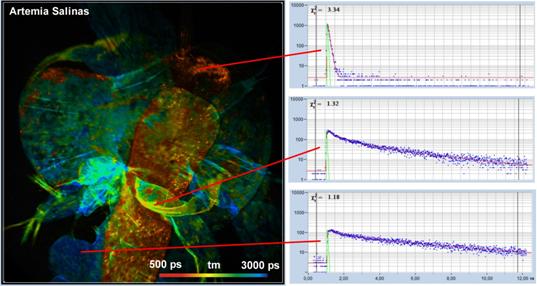
Fig. 55: Two-photon autofluorescence FLIM image of Artemia salinas, mean
(amplitude-weighted) lifetime of double-exponential decay. Decay functions of
selected areas shown on the right.
Metabolic Imaging by NADH FLIM
For several decades, it has been attempted
to obtain metabolic information from the fluorescence lifetime of NADH (nicotinamide
adenine dinucleotide). These attempts were not successful in deriving the
metabolic state because the lifetimes of the NADH decay components depend also
on molecular parameters other than the metabolic state. bh FLIM systems have
overcome the problem by using either the amplitude ratio of the decay
components (a1/a2) or the amplitude of the decay component of free NADH, a1. It
turns out that a1/a2 (the metabolic ratio) or a1 (the metabolic indicator)
describe the metabolic state independently of the type of the cell and its molecular
environment. Tumor cells have an a1 above 0.7, normal cells an a1 below 0.7. An
example is shown in Fig. 56.
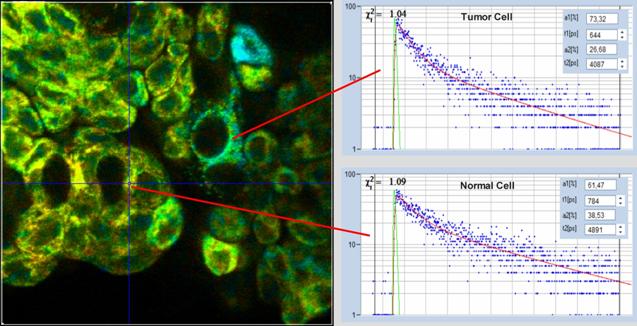
Fig. 56: Metabolic FLIM, a1 image and decay curves. Upper curve: Tumor
cell. Lower curve: Normal cell. The tumor cell has an a1 above 0.7. Live human
bladder cells from a biopsy. Data analysis with SPCImage NG, MLE algorithm.
NADH FLIM for Clinical Diagnostics
It has been shown that NADH FLIM can be
used for clinical diagnostics. FLIM data were obtained from human bladder cells
excised during surgery. The patients had been diagnosed with bladder cancer or
other suspicious lesions by classic endoscopy. Measurements on the excised
material were performed immediately after surgery, before the material went to
histology [49]. By using a normal / cancer discrimination threshold of
a1 = 0.71 perfect agreement with the histology results was obtained.
Please see [1, 19, 20, 49] for
details.
NADH FLIM with Multiphoton Excitation and Ultra-Fast
Detectors
Metabolic FLIM requires double-exponential
data analysis to extract the amplitudes of the decay components of bound and
unbound NADH. Separation of the components improves with the time resolution of
the FLIM system [34]. The DCS-120 MP multiphoton system in combination with the
ultra-fast HPM-100-06 and -07 detectors achieves a time resolution (width of
the instrument-response function) of less than 20 ps FWHM [1, 33]. The fast
response greatly improves the accuracy at which fast decay components can be
extracted from a multi-exponential decay. Date taken with the system not only
show the metabolic state of the cells reliably, they also show heterogeneity in
the a1 of different mitochondria. An NADH FLIM image recorded with the DCS-120
MP and an HPM-100-06 is shown in Fig. 57. The cells were cultured from a
biological cell line. These cells are derived from tumor cells, therefore a1 is
larger than 0.7.
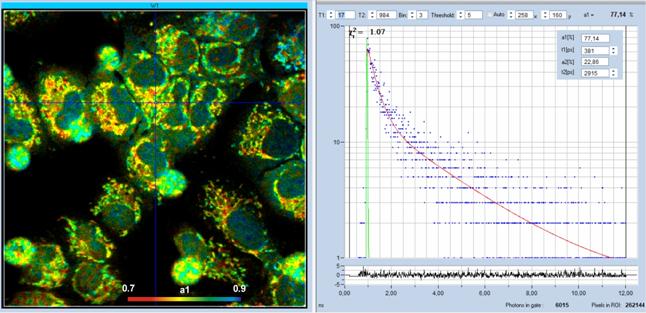
Fig. 57: Left: NADH Lifetime image, amplitude of free NADH, a1. Right:
Decay curve at cursor position, 4x4 pixel area. DCS-120 MP with HPM-100-06
detector, FLIM data format 512 x 512 pixels, 1024 time channels. SPCImage NG
data analysis.
Metabolic Imaging by simultaneous FLIM of NADH and FAD
When it comes to spectroscopic measurement
of the metabolic state often only the fluorescence of NADH is considered.
However, also the fluorescence of FAD (flavin adenine dinucleotide) shows a
dependence on the metabolic state. Like NADH, FAD exists in a bound and an
unbound component. The bound / unbound ratio depends on the metabolic
state. The components have different lifetimes, and can be separated by
double-exponential decay analysis. The amplitudes of the decay components or
the ratio of the amplitudes depend on the metabolic state [2, 1, 46, 50]. Recording FAD FLIM in
combination with NADH FLIM may therefore increase the reliability of metabolic
imaging. However, there is a problem: The excitation of NADH inevitably also
excites FAD, and the fluorescence of FAD cannot be separated from the
fluorescence of NADH by emission filtering. The DCS-120 system therefore uses
excitation-wavelength multiplexing in combination with dual-channel detection [1,
19], see 'Laser Wavelength Multiplexing', page 30. An example is shown in Fig. 58.
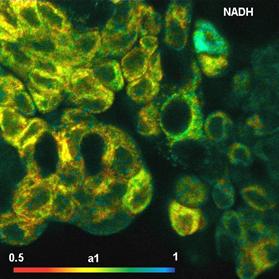
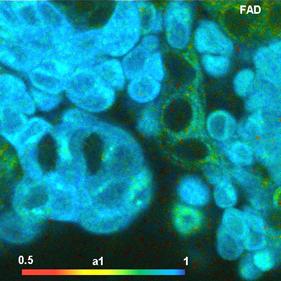

Fig. 58: NADH and FAD images, showing the amplitude of the fast decay
component, a1. Same sample as shown in Fig. 56.
Detection of FMN
A problem of using FAD for metabolic
imaging is that a part of the fluorescence in the FAD emission range comes from
FMN. FNM has a similar emission spectrum as FAD but a different fluorescence
lifetime. FMN does not react to the metabolic state the same was as FAD.
Therefore its presence can be a problem for quantitative metabolic FLIM. With
the high quality of the DCS-120 data and the superior multi-exponential
capabilities of SPCImage NG data analysis FAD and FMN can be distinguished in
the FLIM data. Fig. 59 shows images of fractions of bound FAD (a1), free FAD
(a2), and FMN (a3) in live human bladder cells.
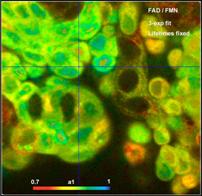
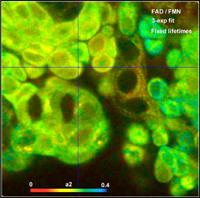
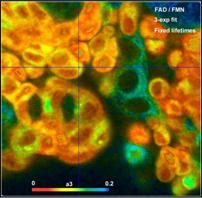
Fig. 59: Amount of bound FAD, free FAD, and FMN in live human bladder
cells. Recorded by DCS-120 confocal system.
Label-Free Imaging of Macroscopic Objects
Metabolic FLIM of macroscopic objects is
possible with the DCS-120 MACRO system. It differs from the other DCS systems
in that the sample is placed directly, without a microscope, in the image plane
of the DCS scanner. Objects as large as 20 mm can be imaged in a single scan [1,
9], see Fig. 5, page 7. Fig. 60 shows an NADH image of open tumor in a mouse.
Decay curve and decay parameters in selected spots are shown on the right. The
metabolic parameter, a1, is 0.61 in the healthy tissue and 0.84 in the tumor.
This corresponds well with a1 values from other metabolic FLIM data: a1 is
<0.7 in the good tissue and >0.7 in the tumor.
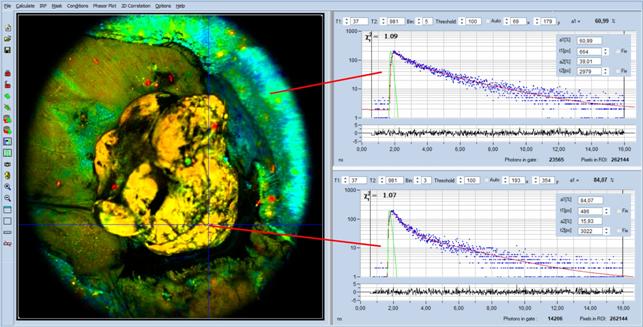
Fig. 60: Open tumor in a mouse, image recorded by DCS-120 MACRO system.
Excitation wavelength 370 nm, detection from420nm to 480 nm. The
metabolic parameter, a1, is <0.7 in the good tissue and >0.7 in the
tumor. This is exactly what is to be expected from other metabolic FLIM
experiments.
Ultra-Fast Fluorescence Decay in Biological Material
Due to the short pulse width of the
femtosecond lasers the DCS-120 MP system delivers extremely high temporal
resolution. An example is shown in Fig. 61. The image shows mushroom spores of Boletus
edulis [23]. The image was recorded by a DCS-120 MP with a Femto-Fibre-Pro
laser (Toptica), ultra-fast HPM-100-06 detectors, and SPC-150NX TCSPC modules.
The data show clearly a decay component of 20 ps lifetime.
An interesting application of ultra-fast
FLIM is lifetime imaging of Carotenoids. The lifetimes are in the lower ps
range but can easily be resolved by the DCS-120 MP system with ultra-fast detectors
[24]. Not only can carotenoids can be localised by their short fluorescence
lifetimes, different carotenoids can also be distinguished by different
lifetime. Moreover, there are indications that similar carotenoids display
different lifetime in different molecular environment. Examples are shown in Fig.
62 and Fig. 63.
Ultra-fast fluorescence decay was also
found in human hair and - interestingly - in malignant melanoma. Please see [25]
and [26].
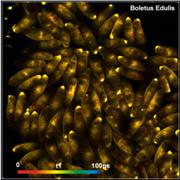
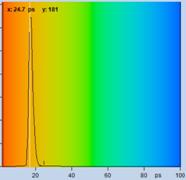
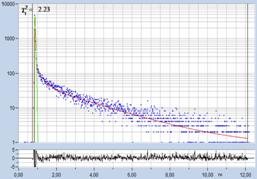
Fig. 61: Spores of Boletus edulis. Left to right: Image of fast
decay component, t1, of triple-exponential decay model, histogram of
t1, and decay curve in selected spot. Data from [23].
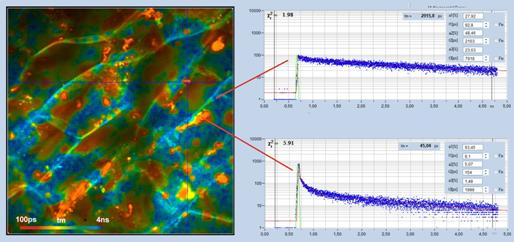
Fig. 62: Lifetime image of carrot tissue. Amplitude-weighted lifetime, tm,
of triple-exponential fit. Decay curves in locations without and with b-carotene shown
on the right. Triple-exponential decay analysis with SPCImage NG. The fast
lifetime component in carotene-rich regions is 8.1 ps.
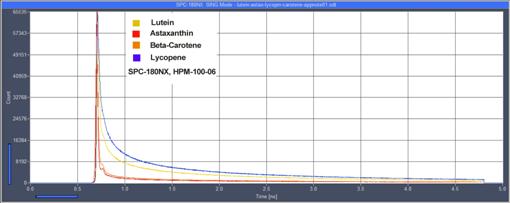
Fig. 63:
Decay curves of lutein, astaxanthin, b-carotene, and Lypcopene. Lifetimes are in the
range from 10 to 20 ps [24].
Oxygen sensing is based on the quenching of
the phosphorescence decay of (exogenous) phosphorescent dyes by oxygen. PLIM
can be recorded by the DCS-120 system using triggered MCS recording and
multi-pulse excitation [32, 1, 39, 41], see also page 31 of this brochure. Two
examples are shown in Fig. 64. The figure shows PLIM images of cultured human
embryonic kidney cells incubated with a palladium-based phosphorescence dye. Fig.
64, left was recorded under atmospheric oxygen partial pressure. The maximum of
the lifetime distribution over the pixels (upper right) is at 75 µs. Fig. 64,
right, was recorded under decreased oxygen partial pressure. As can be seen,
the maximum of the lifetime distribution has shifted to 144 µs.
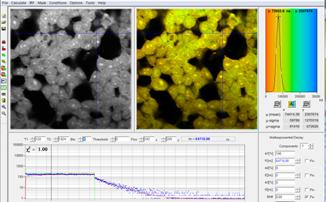
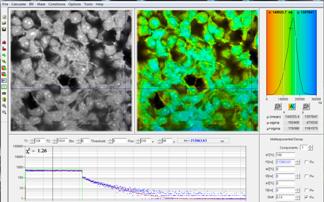
Fig. 64: HEK cells incubated with a palladium dye imaged under different
oxygen partial pressure. Left: Atmospheric O2 pressure. Right: reduced oxygen
partial pressure. Recorded by bh DCS‑120 confocal scanning system, data
analysis by bh SPCImage. Lifetime scale 0 (red) to 300 µs (blue).
Phosphorescence lifetime at the Cursor-Position 65 µs. The maximum of the
lifetime distribution over the pixels is at 75 µs.
There is currently an increasing interest
in PLIM not only for oxygen sensing but also for background-free imaging and
luminescence decay of inorganic compounds. In all these applications the bh
technique delivers a far better sensitivity than PLIM techniques based on
single-pulse excitation.
Simultaneous Sensing of Oxygen and Metabolic State
Oxygen concentration has a large influence
of the metabolism of a cell [42, 43]. In fact, the normal / tumor threshold
of 0.7 for the metabolic indicator, a1, strictly applies only for oxygen
concentrations in the normal physiological range. Consequently, there is large
interest to obtain images of the oxygen concentration and the metabolic state
simultaneously. This is exactly what the bh FLIM / PLIM technique has been
designed for. The technique is based on modulating a ps diode laser synchronously
with the pixel clock of the scanner [1, 32]. FLIM is recorded during the On
time, PLIM during the Off time of the laser. The SPCM software delivers
separate images for the fluorescence and the phosphorescence which are then
analysed with SPCImage NG FLIM/PLIM analysis software [4]. An example is
shown in Fig. 65.
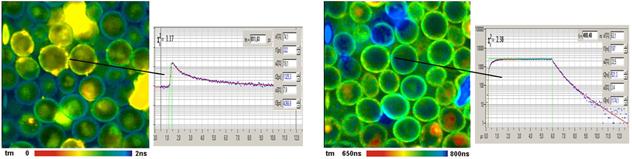
Fig. 65: Yeast cells stained with (2,2-bipyridyl) dichlororuthenium (II)
hexahydrate. FLIM and PLIM image, decay curves in selected spots.
64-bit Data Acquisition Software
The DCS‑120 FLIM systems use the bh
SPCM 64 data acquisition software. SPCM runs the data acquisition in the
various operation modes of the SPC modules while controlling peripheral
devices, such as detectors, lasers, scanners, or motor stages. Operation modes
are available for almost any conceivable TCSPC application. There are modes for
fluorescence and phosphorescence decay recording, multi-wavelength decay
recording, laser-wavelength multiplexing, recording of time series, FCS and
photon counting histograms, and there are modes for FLIM, multi-wavelength
FLIM, Mosaic FLIM, time-series FLIM, Z stack FLIM, and simultaneous FLIM/PLIM.
Since July 2019 SPCM comes with extended multi-threading capabilities, greatly
improving the throughput rate even in case of complex online data and display
operations. A direct link is provided for communication with SPCImage NG FLIM
analysis software.
Since 2013 the SPCM software is available
in a 64-bit version. SPCM 64 bit exploits the full capability of Windows 64
bit, resulting in faster data processing, capability of recording images of extremely
large pixel numbers, and availability of additional multi-dimensional FLIM
modes [1, 31].
The Main Panel of SPCM is configurable by
the user [1]. Different configurations can thus be created for different
applications and measurement tasks. The configurations can be stored in a
Predefined Setup panel and recalled on demand by a single mouse click. A few typical configurations for FLIM systems are shown in Fig. 66. A
154-page description of the SPCM software is available in [1].
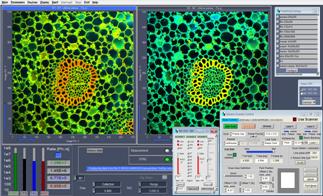
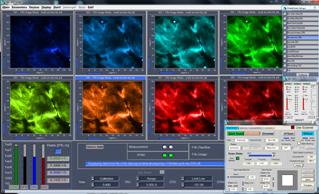
Fig. 66: Main panel of SPCM software. Configurations for dual-channel FLIM,
multi-wavelength FLIM, single-channel MCARO FLIM, multi-wavelength curve mode.
Easy Change Between Instrument Configurations
Frequently used instrument configurations
are stored in a Predefined Setup panel. Changing between the different
configurations and user interfaces is just a matter of a single mouse click,
see Fig. 67.
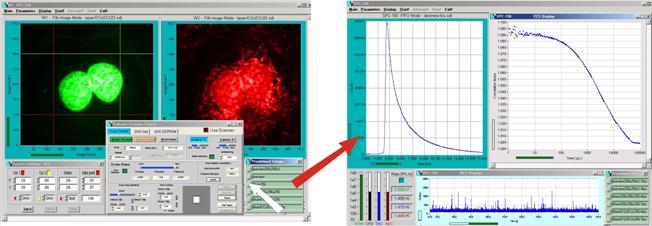
Fig. 67: Changing between different instrument configurations: The DCS-120
system switches from a FLIM configuration into an FCS configuration by a simple
mouse click
Integrated Scanner Operation
Scanner control is fully integrated in the
data acquisition software. It includes definition of the frame size, pixel
numbers, scan speed, and zoom factor. A fast preview mode is provided for
sample setup and focus tuning. For a detailed description please see [1] and [2].
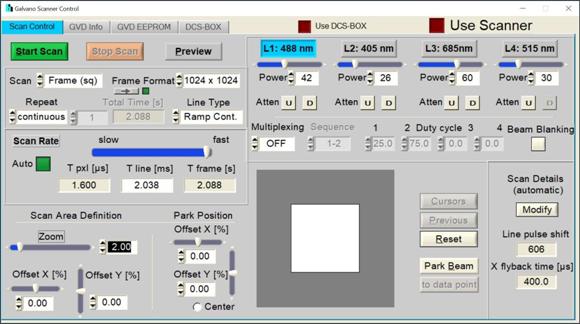
Fig. 68: Scanner Control Panel of the
DCS-120 system
Interactive Scanner Control
The zoom factor and the position of the
scan area can be adjusted via the scanner control panel or via the cursors of
the display window. Changes in the scan parameters are executed online, without
stopping the scan. The result becomes immediately visible in the preview images.
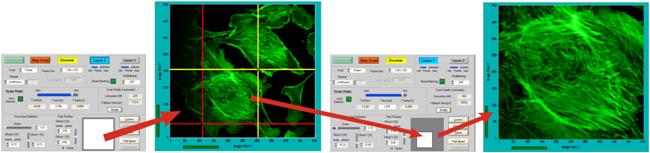
Fig. 69:
Interactive scanner control
Automatic Scanner Speed
The DCS-120 scanner control automatically
selects the maximum speed of the scanner. Unless otherwise selected, the
scanner thus always runs at the highest possible pixel rate, resulting in fast
acquisition, minimum triplet excitation, and minimum photobleaching.
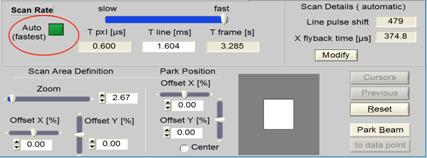
Fig. 70: Automatic selection of scan
speed
Integrated Laser Control
Control of up to four lasers is implemented
in the scanner control panel. It includes selection of an active laser, beam
blanking during the line and frame flyback, intensity control, and laser
multiplexing.
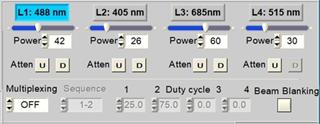
Fig. 71: Laser control part of the
scan control panel
Integrated Control of Peripheral Devices
Control of peripheral devices, such as
motorised scan stages, Ti:Sa lasers, AOMs for Ti:Sa lasers, or Z drives of
microscopes is integrated in the DCS operating software.
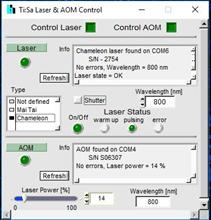
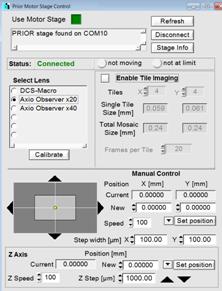
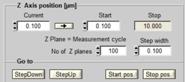
Fig. 72: Control of peripheral
devices. Ti:Sa laser and AOM, motorised sample stage, Z drive of Axio Observer
The DCS-120 system
uses SPCImage NG FLIM data analysis. SPCImage NG is a new generation of bh's legendary
SPCImage TCSPC-FLIM data analysis software. It combines time-domain and
frequency-domain analysis, uses a maximum-likelihood (MLE) algorithm to
calculate the parameters of the decay functions in the individual pixels, and
accelerates the analysis procedure by GPU processing. 1D and 2D parameter
histograms are available to display the distribution of the decay parameters
over the pixels of the image or over selectable ROIs. Image segmentation can be
performed via the phasor plot. Pixels with similar phasor signature can be
combined for high-accuracy time-domain analysis. SPCImage NG provides decay
models with one, two, or three exponential components, incomplete-decay models,
and shifted-component models. Another important feature is advanced IRF
modelling, making it unnecessary to record IRFs for the individual FLIM data
sets. For a detailed description please see [1, 2, 4], and SPCImage NG Overview Brochure [5]. A typical main panel of SPCImage NG is
shown in Fig. 73.
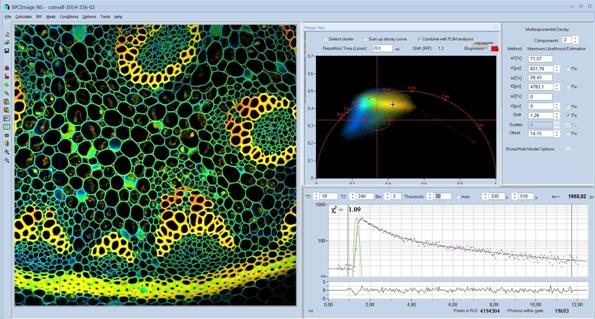
Fig. 73: Example of SPCImage NG main panel. Combination of time-domain analysis (left and lower
right) and phasor plot (upper right)
Deconvolution and Fit Procedure
SPCImage NG runs
an iterative fit and de-convolution procedure on the decay data of the individual
pixels of the FLIM images. In the simplest case, the result is the lifetime of
the decay functions in the individual pixels. For complex decay functions the
fit procedure delivers the lifetimes and amplitudes of the decay components.
SPCImage then creates colour-coded images of the amplitude- or
intensity-weighted lifetimes in the pixels, images of the lifetimes or
amplitudes of the decay components, images of lifetime or amplitude ratios, and
images of other combinations of decay parameters, such as FRET intensities,
FRET distances, bound-unbound ratios, or the fluorescence-lifetime redox ratio,
FLIRR. A few examples are shown in Fig. 74 and Fig. 75. For details please see
[1, 2, 4].
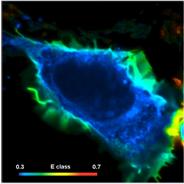
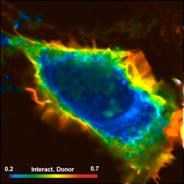
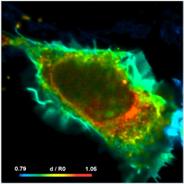
Fig. 74: Cell with interacting proteins, labelled with a FRET donor and a
FRET acceptor. Left to right: Classic FRET efficiency, fraction of interacting
donor, FRET distance
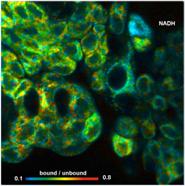
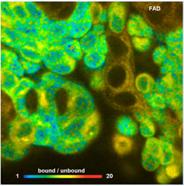
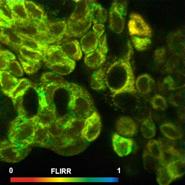
Fig. 75: Metabolic FLIM. Bound-unbound ratio of NADH, Bound/unbound ratio
of FAD, Fluorescence-Lifetime Redox Ratio, FLIRR.
GPU Processing
SPCImage NG uses GPU (Graphics Processor Unit)
processing. GPU processing is running on NVIDIA cards and a number of other
NVIDIA-compatible devices. The TCSPC data are transferred into the GPU, which
then runs the de-convolution and fit procedure for a large number of pixels in
parallel. This way, data processing times for large images are reduced from
formerly more than 10 minutes to a few seconds.
Maximum-Likelihood Algorithm
Unlike earlier
SPCImage versions, SPCImage NG uses a maximum-likelihood algorithm (or
maximum-likelihood estimation, MLE) for fitting the data. In contrast to the
usual least-square fit, the MLE algorithm takes into account the Poissonian
distribution of the photon numbers. Compared to the least-square method, the
fit accuracy is improved especially for low photon numbers, and there is no
bias toward shorter lifetime as it is often observed for the least-square fit.
For comparison with older data sets the weighted least-square fit and the
first-moment algorithms of the previous SPCImage versions are still available
in SPCImage NG.
Instrument-Response Function
SPCImage NG
avoids troublesome recording of an instrument response function (IRF) for each
FLIM measurement. This is achieved by modelling the IRF with a generic function.
The parameters of this function are determined by fitting it to the FLIM data
together with the selected decay model. The results of this procedure are so
good that an accurate IRF is obtained even for decay functions containing
ultra-fast components, see Fig. 76. For details please see [4].
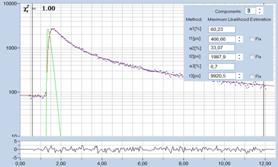
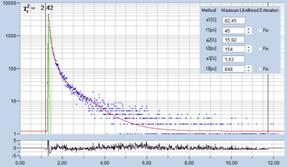
Fig. 76: Synthetic IRF. Left: Autofluorescence of cells, ps diode laser,
HPM-100-40. Right: Sample with extremely fast decay component, femtosecond
fibre laser, HPM-100-06
Phasor Plot
SPCImage NG combines time-domain
multi-exponential decay analysis with a phasor plot. In the phasor plot, the
decay data in the individual pixels are expressed as phase and amplitude values
in a polar diagram [38].
Independently of their location in the image, pixels with similar decay
signature form clusters in the phasor plot. Different phasor clusters can be selected,
and the corresponding pixels back-annotated in the time-domain FLIM images. The
decay functions of the pixels within the selected phasor range can be combined
into a single decay curve of high photon number. This curve can be analysed at
high accuracy, revealing decay components that are not visible by normal
pixel-by pixel analysis [4, 35]. An example is shown in Fig. 77.
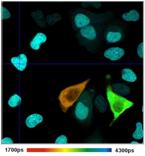
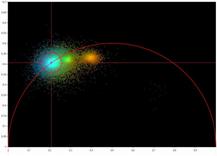
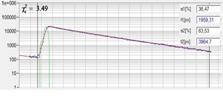
Fig. 77: Combination of time-domain analysis with phasor plot. Left to
right: Lifetime image, phasor plot, decay curve of combined pixels within
selected phasor range
Image Segmentation
SPCImage NG provides automatic image segmentation
functions via the phasor plot and 2D histograms of the decay parameters. Areas
with different decay signature form separate clusters in these presentations.
Interesting clusters can be selected and back-annotated in the images. The
decay data of the corresponding pixels are combined into a single decay curve
with extremely high photon number. Multi-exponential decay analysis on the combined
data delivers precision decay parameters even if the photon numbers in the
individual pixels are low. An example is shown in Fig. 78.
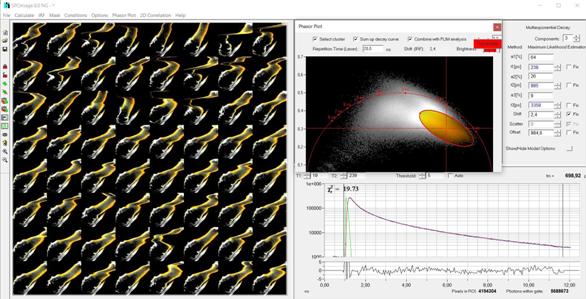
Fig. 78: Image segmentation on temporal-mosaic FLIM data of a live water
flee
Single-Curve Analysis
SPCImage can also be used to analyse single
decay curves, The data can come from traditional cuvette experiments, or from
the combined pixels of a FLIM recording. An example is shown in Fig. 79.
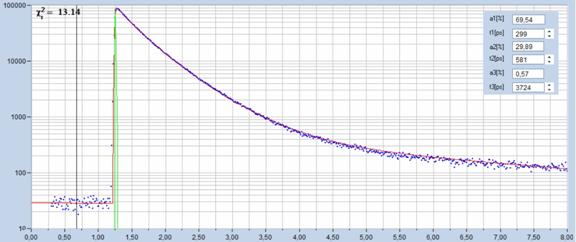
Fig. 79: SPCImage used for fluorescence decay analysis of single curves.
NADH dissolved in water, recorded with bh DCS-120 MP FLIM system.
Summary
The DCS-120 is a high-performance lifetime
imaging system based on laser scanning and multi-dimensional TCSPC. The system
is characterised by high time resolution, high sensitivity, high photon
efficiency, high spatial resolution, and suppression of out-of-focus
fluorescence and laterally and longitudinally scattered light. The system comes
in different versions: The standard DCS-120 system uses excitation by ps diode
lasers and confocal detection, the DCS-120 MP systems use multiphoton
excitation and non-descanned detection. The DCS-120 MP Fibre uses a
femtosecond fibre laser for excitation, making it the cheapest multiphoton
microscope on the market. All systems can be delivered as complete laser
scanning microscopes or as FLIM upgrades for existing conventional microscopes.
Moreover, there is the DCS-120 MACRO system for scanning cm-size objects directly
in the primary image plane of the scanner.
The DCS system can be used for the whole
range of FLIM applications: From entry-level to high end molecular imaging. The
DCS-120 is based on a new understanding of FLIM in general.
Different than other FLIM techniques and
FLIM systems which consider FLIM just a way to improve contrast in laser
scanning microscopy, the DCS-120 has been designed with molecular-imaging
applications in mind. It thus has capabilities beyond the reach of other
systems: Compatibility with live-cell imaging, extraordinarily high time
resolution and photon efficiency, capability to split decay functions into
several components, excitation-wavelength multiplexing in combination with
parallel-channel detection, recording of dynamic lifetime effects caused by
fast physiological effects, and simultaneous FLIM/PLIM. Typical applications
are measurement of molecular-environment parameters, protein-interaction
experiments by FRET techniques, label-free imaging, imaging of the metabolic
state of cells and tissues, the use of endogenous fluorophores with lifetimes
in the 10-ps range, oxygen-concentration measurement, and recording of fast
physiological processes in biological systems. No other FLIM technique and no
other FLIM system offers a similar range of advanced capabilities.
Scan head bh
DCS-120 scan head
Optical principle confocal,
beam scanning by fast galvanometer mirrors
Laser inputs two
independent inputs, fibre coupled or free beam
Laser power regulation, optical continuously
variable via neutral-density filter wheels
Outputs to detectors two
outputs, detectors are directly attached
Main beamsplitter versions multi-band
dichroic, wideband, multiphoton
Secondary beamsplitter wheel 3 dichroic
beamsplitters, polarising beamsplitter, 100% to channel1, 100% to channel2
Pinholes independent
pinhole wheel for each channel
Pinhole size 11
pinholes, from about 0.5 to 10 AU
Emission filters 2
filter sliders per channel
Connection to microscope adapter
to left side port or port on top of microscope
Coupling of lasers into scan head (visible)
single-mode fibres, Point-Source type, separate for each laser
Coupling of laser into scan head (Ti:Sa) free
beam, 1 to 2 mm diameter
Scan Controller bh
GVD-120
Principle Digital
waveform generation, scan waveforms generated by hardware
Scan waveform linear
ramp with cycloid flyback
Scan format line,
frame, or single point
Frame size, frame scan 16x16
to 4096x4096 pixels
line scan 16
to 4096 pixels
Laser power control, electrical via
electrical signal to lasers
Laser multiplexing frame
by frame, line by line, or within one pixel
Beam blanking during
flyback and when scan is stopped
Scan rate automatic
selection of fastest rate or manual selection
minimum pixel time for frame size 64x64 128x128 256x256 512x512 1024x1024 2048x2048
Zoom=1 25.6µs 12.8µs 6.4µs 3.2µs 1.6µs 1.2µs
Zoom=8 6.4µs 3.2µs 1.6µs 0.8µs 0.6µs 0.5µs
minimum frame time for frame size 64x64 128x128 256x256 512x512 1024x1024 2048x2048
Zoom=1 0.19s 0.37s 0.64s 1.24s 2.6s 6.5s
Zoom=8 0.037s 0.074s 0.173s 0.320s 1.0s 2.7s
Scan area definition via
zoom and offset or interactive via cursors during preview
Fast preview function 1
second per frame, 128 x 128 pixels
Beam park function via
cursor in preview image or cursor in FLIM image
Laser control 2
Lasers or 4 Lasers, on/off, frame, line, pxl multiplexing
Diode lasers BDS-SMN laser
Number of lasers simultaneously operated 2
Wavelengths 375nm,
405nm, 445nm, 473nm, 488nm, 510nm, 640nm, 685nm, 785nm
Pulse width, typical 30
to 70 ps
Pulse frequency 20MHz,
50MHz, 80MHz, CW
Power in picosecond mode 0.25mW
to 1mW injected into fibre. Depends on wavelength version.
Power in CW mode 10
to 40mW injected into fibre. Depends on wavelength version.
Other lasers
Visible and UV range any
ps pulsed laser of 20 to 80 MHz repetition rate
Coupling requirements Point-Source
Kineflex compatible fibre adapter
Wavelength any
wavelength from 400nm to 800nm
fs NIR Lasers for multiphoton operation any
fs laser
Coupling requirements free
beam, diameter 1 to 2 mm
Wavelength 700
to 1200 nm
Detectors (standard) bh
HPM-100-40 hybrid detector
Spectral Range, incl. DCS optics 330
to 710nm
Peak quantum efficiency 40
to 50%
System IRF width with bh diode laser 120
to 130 ps
System IRF with fs laser 90
to 100 ps
Background count rate, thermal 300
to 2000 counts per second
Power supply, gain control, overload
shutdown via DCC-100 controller of TCSPC system
Detectors (optional) bh
HPM-100-06 and HPM-100-07 hybrid detectors
Spectral Range, incl. DCS optics 330 nm to 600 nm 330
to 850 nm
Peak quantum efficiency 20
% (at 400nm) 26% at 290 nm, 22% at 400nm
System IRF width with fs Ti:Sa laser <20
ps
System IRF width with bh ps diode laser 38
to 90 ps
Active area 3mm
Background count rate, thermal 100
to 1000 counts per second
Power supply, gain control, overload
shutdown via DCC-100 controller of TCSPC system
Detectors (optional) bh
HPM-100-50 hybrid detector
Spectral Range 400
to 900nm
Peak quantum efficiency 12
to 15%
IRF width with bh diode laser 150
to 220 ps
Active area 3mm
Background count rate, thermal 1000
to 8000 counts per second
Power supply, gain control, overload
shutdown via DCC-100 controller of TCSPC system
Detectors (optional) bh
MW FLIM GaAsP Multi-Wavelength FLIM detector
Spectral range 380
to 700nm
Number of wavelength channels 16
Spectral width of wavelength channels 12.5
nm
IRF width with bh diode laser 200
to 250 ps
Power supply and overload shutdown via
DCC-100 controller of TCSPC system
TCSPC System Two
bh SPC-180N modules or One SPC-QC-104 module [1]
Number of modules (recording channels) 2 2
(3 with additional detector)
Electrical time resolution 3 ps
fwhm 38 ps (SPC-QC-104)
Minimum time channel width 813
fs 4 ps
Dead time 80 ns 10 ns
Saturated count rate 10 MHz
per channel 100 MHz per channel
Input from detector constant-fraction
discriminator
Reference (SYNC) input constant-fraction
discriminator
Synchronisation with scanning via
frame clock, line clock and pixel clock pulses
Scan rate any
scan rate
Synchronisation with laser multiplexing via
routing function
Recording of multi-wavelength data simultaneous,
via routing function
Basic acquisition principles on-board-buildup
of photon distributions
buildup
of photon distributions in computer memory
generation
of parameter-tagged single-photon data
online
auto or cross correlation and PCH
Operation modes f(t),
oscilloscope, f(txy), f(t,T), f(t) continuous flow
FIFO
(correlation / FCS / MCS) mode
FIFO
imaging, with MCS imaging, mosaic imaging, time-series imaging
Multi-detector
operation, laser multiplexing operation
cycle
and repeat function, autosave function
Max. Image size, pixels (SPCM 64 bit software) 2048x2048 1024x1024 512x512
No of time channels, see [1] 256 1024 4096
Data Acquisition Software, please see [1]
for details
Operating system Windows
10 or Windows 11, 64 bit
Loading of system configuration single
click in predefined setup panel
Start / stop of measurement by
operator or by timer, starts with start of scan, stops with end of frame
Online calculation and display, FLIM,
PLIM in intervals of Display Time, min. 1 second
Online calculation and display, FCS,
PCH in intervals of Display Time, min. 1 second
Number of images diplayed
simultaneously max 8
Number of curves (Decay, FCS, PCH,
Multiscaler) 16 in one curve window
Cycle, repeat, autosave functions user-defined,
used for
for
time-series recording, Z stack FLIM,
microscope-controlled
time series
Saving of measurement data User
command or autosave function
Optional
saving of parameter-tagged single-photon data
Link to SPCImage data analysis automatically
after end of measurement or by user command
References
1.
W. Becker, The bh TCSPC Handbook. 10th edition. Becker
& Hickl GmbH (2023), available on www.becker-hickl.com, printed copies available from bh
2.
Becker & Hickl GmbH, DCS-120 Confocal and
Multiphoton FLIM Systems, user handbook, 9th edition 2021. www.becker-hickl.com, printed copies available
3.
W. Becker, Advanced time-correlated single-photon counting techniques. Springer,
Berlin, Heidelberg, New York, 2005
4. SPCImage NG next generation FLIM data analysis software. In: W.
Becker, The bh TCSPC Handbook, 10th ed., (2023). Available on
www.becker-hickl.com.
5. Becker & Hickl GmbH, SPCImage NG next generation FLIM data
analysis software. Overview brochure, available on www.becker-hickl.com
6. W. Becker, Bigger and Better Photons: The Road to Great FLIM
Results. Education brochure, available on www.becker-hickl.com.
7. W. Becker, Introduction to Multi-Dimensional TCSPC.
In W. Becker (ed.) Advanced time-correlated single photon counting
applications. Springer, Berlin, Heidelberg, New York (2015)
8.
W. Becker, V. Shcheslavskiy, H. Studier, TCSPC FLIM with Different
Optical Scanning Techniques, in W. Becker (ed.) Advanced time-correlated
single photon counting applications. Springer, Berlin, Heidelberg, New York
(2015)
9. W. Becker, L. Braun, J. Heitz, V. Shcheslavskiy, M. Shirmanova,
Metabolic FLIM of Macroscopic Objects, Application note, available from
www.becker-hickl.com.
10. Wolfgang Becker, Julius Heitz, Lukas Braun, Axel Bergmann, High
Resolution Z-Stack FLIM with the Becker & Hickl DCS‑120 Confocal FLIM
System. Becker & Hickl GmbH, Application note, available on
www.becker-hickl.com
11. W. Becker, H. Netz, DCS-120 Confocal Scanning FLIM System:
Two-Photon Excitation with Non-Descanned Detection. Application note, available
on www.becker-hickl.com
12. W. Becker, C. Junghans, L. Braun, A. Jelzow, Two-Photon FLIM with a
Small Femtosecond Fibre Laser. Application note, available on
www.becker-hickl.com
13. W. Becker, A. Bergmann, C. Biskup, T. Zimmer, N. Klöcker, K. Benndorf, Multi-wavelength TCSPC
lifetime imaging, Proc. SPIE 4620 79-84 (2002)
14. W. Becker, A. Bergmann, M.A. Hink, K. König, K. Benndorf, C. Biskup,
Fluorescence lifetime imaging by time-correlated single photon counting, Micr.
Res. Techn. 63, 58-66 (2004)
15. W. Becker, A. Bergmann, C. Biskup, Multi-Spectral Fluorescence
Lifetime Imaging by TCSPC. Micr. Res. Tech. 70, 403-409 (2007)
16. W. Becker, B. Su, K. Weisshart, O .Holub, FLIM and FCS Detection in
Laser-Scanning Microscopes: Increased Efficiency by GaAsP Hybrid Detectors.
Micr. Res. Tech. 74, 804-811 (2011)
17. W. Becker, Fluorescence Lifetime Imaging - Techniques and
Applications. J. Microsc. 247 (2) (2012)
18. W. Becker, V. Shcheslavkiy, S. Frere, I. Slutsky, Spatially Resolved
Recording of Transient Fluorescence-Lifetime Effects by Line-Scanning TCSPC.
Microsc. Res. Techn. 77, 216-224 (2014)
19. W. Becker, A. Bergmann, L. Braun, Metabolic Imaging with the DCS-120
Confocal FLIM System: Simultaneous FLIM of NAD(P)H and FAD, Application note,
available on www.becker-hickl.com (2018)
20. W. Becker, R. Suarez-Ibarrola, A. Miernik, L. Braun, Metabolic
Imaging by Simultaneous FLIM of NAD(P)H and FAD. Current Directions in
Biomedical Engineering 5(1), 1-3 (2019)
21. W. Becker, A Common Mistake in Lifetime-Based FRET Measurements.
Application note, Becker & Hickl GmbH (2023)
22. W. Becker, A. Bergmann, Double-Exponential FLIM-FRET Approach is
Free of Calibration. Application note, Becker & Hickl GmbH (2023)
23. W. Becker, C. Junghans, A. Bergmann, Two-Photon FLIM of Mushroom
Spores Reveals Ultra-Fast Decay Component. Application note (2021), available
on www.becker-hickl.com.
24. W. Becker, A. Bergmann, C. Junghans, Ultra-Fast Fluorescence Decay
in Natural Carotenoids. Application note, www. becker-hickl.com (2022)
25. W. Becker, C. Junghans, V. Shcheslavskiy, High-Resolution
Multiphoton FLIM Reveals Ultra-Fast Fluorescence Decay in Human Hair.
Application note, www. becker-hickl.com (2023)
26. W. Becker,V. Shcheslavskiy, V. Elagin, Ultra-Fast Fluorescence Decay
in Malignant Melanoma. Application note, available on www. becker-hickl.com
27. Becker & Hickl GmbH, The HPM‑100-40 hybrid detector.
Application note, available on www.becker-hickl.com
28. Becker & Hickl GmbH, Spatially resolved recording of
fluorescence-lifetime transients by line-scanning TCSPC. Application note,
available on www.becker-hickl.com
29. Becker & Hickl GmbH, DCS-120 Confocal Scanning FLIM System with
User-Specific Lasers: FIANIUM SC400 Laser. Application note, available on
www.becker-hickl.com
30. Becker & Hickl GmbH, DCS-120 Confocal Scanning FLIM System with
User-Specific Lasers: NKT SuperK EXTREME Laser. Application note, available on
www.becker-hickl.com
31. Becker & Hickl GmbH, Megapixel FLIM with bh TCSPC Modules -
The New SPCM 64-bit Software. Application note, available on www.becker-hickl.com
32. Becker & Hickl GmbH, Simultaneous Phosphorescence and
Fluorescence Lifetime Imaging by Multi-Dimensional TCSPC and Multi-Pulse
Excitation. Application note, available on www.becker-hickl.com
33. Becker & Hickl GmbH, Sub-20ps IRF Width from Hybrid
Detectors and MCP-PMTs. Application note, available on www.becker-hickl.com
34. Becker & Hickl GmbH, Ultra-fast HPM Detectors Improve
NAD(P)H FLIM. Application note, available on www.becker-hickl.com
35. Becker & Hickl GmbH, New SPCImage Version Combines Time-Domain
Analysis with Phasor Plot. Application note, available on www.becker-hickl.com
36. Becker & Hickl GmbH, Two-Photon FLIM with a Femtosecond Fibre
Laser. Application note, available on www.becker-hickl.com
37. D. Chorvat, A. Chorvatova, Multi-wavelength fluorescence lifetime
spectroscopy: a new approach to the study of endogenous fluorescence in living
cells and tissues. Laser Phys. Lett. 6 175-193 (2009)
38. M. A. Digman, V. R. Caiolfa, M. Zamai, and E. Gratton, The phasor
approach to fluorescence lifetime imaging analysis, Biophys J 94, L14-L16
(2008)
39. R. I. Dmitriev, S. M. Borisov, A. V. Kondrashina, J. M. P. Pakan, U.
Anilkumar, J. H. M. Prehn, A. V. Zhdanov, K. W. McDermot, I. Klimant, D. B.
Papkovsky, Imaging oxygen in neural cell and tissue models by means of anionic
cell-permeable phosphorescent nanoparticles. Biomaterials 34, 9307-9317 (2013)
40. S. Frere, I. Slutsky, Calcium imaging using Transient Fluorescence-Lifetime Imaging
by Line-Scanning TCSPC. In: W. Becker (ed.) Advanced time-correlated single
photon counting applications. Springer, Berlin, Heidelberg, New York (2015)
41. J. Jenkins, R. I. Dmitriev, D. B. Papkovsky, Imaging Cell and Tissue O2 by TCSPC-PLIM. In: W. Becker (ed.) Advanced
time-correlated single photon counting applications. Springer, Berlin,
Heidelberg, New York (2015)
42. S. Kalinina, V. Shcheslavskiy, W. Becker, J. Breymayer, P. Schäfer,
A. Rück, Correlative NAD(P)H-FLIM and oxygen sensing-PLIM for metabolic
mapping. J. Biophotonics 9(8):800-811 (2016)
43. H. Kurokawa, H. Ito, M. Inoue, K. Tabata, Y. Sato, K. Yamagata, S.
Kizaka-Kondoh, T. Kadonosono, S. Yano, M. Inoue & T. Kamachi, High
resolution imaging of intracellular oxygen concentration by phosphorescence
lifetime, Scientific Reports 5, 1-13 (2015)
44. J.R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn.,
Springer (2006)
45. X. Liu, D. Lin, W. Becker, J. Niu, B. Yu, L. Liu, J. Qu, Fast
fluorescence lifetime imaging techniques: A review on challenge and
development. Journal of Innovative Optical Health Sciences, 1930003-1 to -27 (2019)
46. M. M. Lukina, V. V. Dudenkova, N. I. Ignatovaa, I. N. Druzhkova, L.
E. Shimolina, E. V. Zagaynovaa, M. V. Shirmanova, Metabolic cofactors NAD(P)H
and FAD as potential indicators of cancer cell response to chemotherapy with
paclitaxel. BBA General Subjects 1862, 1693-1700 (2018)
47. A. Rück, C. Hauser, S. Mosch, S. Kalinina, Spectrally resolved
fluorescence lifetime imaging to investigate cell metabolism in malignant and
nonmalignant oral mucosa cells. J. Biomed. Opt. 19(9), 096005-1 to -9 (2014)
48. T. F. Sergeeva, M. V. Shirmanova, O. A. Zlobovskaya, A. I. Gavrina,
V. V. Dudenkova, M. M. Lukina, K. A. Lukyanov, E. V.
Zagaynova, Relationship between intracellular pH, metabolic cofactors, and caspase-3 activation in cancer cells during apoptosis. BBA - Molecular
Cell Research (2017)
49. R. Suarez-Ibarrola, L. Braun, P. Fabian Pohlmann, W. Becker, A.
Bergmann, C. Gratzke, A. Miernik, K. Wilhelm, Metabolic Imaging of Urothelial
Carcinoma by Simultaneous Autofluorescence Lifetime Imaging (FLIM) of NAD(P)H
and FAD. Clinical Genitourinary Cancer (2020)
50. A. J. Walsh, A. T. Shah, J. T. Sharick, M. C. Skala, Fluorescence Lifetime
measurements of NADH in live cells and tissue. In: W. Becker (ed.) Advanced
time-correlated single photon counting applications. Springer, Berlin,
Heidelberg, New York (2015)
