DCS-120 Confocal
Scanning FLIM System: Two-Photon Excitation with Non-Descanned Detection
Wolfgang Becker, Holger Netz, Becker & Hickl GmbH,
Berlin, Germany
Abstract: This application note shows how the bh DCS‑120 confocal FLIM
system can be upgraded for multiphoton FLIM with non-descanned detectors. With
the upgraded system, highly detailed lifetime images from unstained mammalian
skin could be recorded from as deep as 90 µm below the surface. The
efficiency of the system is excellent. An average count rate of 1 MHz
could be obtained without detectable photobleaching within an acquisition time
of two minutes. The quality of the data obtained within this time was adequate
for triple-exponential decay analysis.
Two-Photon Excitation
Simultaneous absorption of two (or more) photons
has been theoretically predicted by Maria Göppert-Mayer [7]. It has been suggested for laser scanning
microscopy by Wilson and Sheppard in 1984 [8] and practically introduced by W.
Denk and J.H. Strickler [6] in 1990. The efficiency of two-photon excitation
increases with the square of the excitation power density. If a femtosecond Titanium-Sapphire
laser is focused into a diffraction-limited volume by a microscope lens the
excitation process becomes so efficient that no more than a about ten milliwatt
of laser power are required to obtain clear fluorescence images with endogenous
and exogenous fluorophores.
Two photon excitation has a number of advantages
over one-photon excitation.
First, the absorption of the sample at the
NIR wavelength of the Ti:Sa laser is low. Also the scattering coefficient is
lower than at visible or ultraviolet wavelengths. Two-photon excitation
therefore goes deeper into tissue than one-photon excitation, see Fig. 1, left
and middle. Two-photon excitation can be used to excite fluorescence in tissue
layers as deep as 100 um, in some cases even 1 mm.
Second, noticeable two-photon excitation is
obtained only in the focus of the microscope lens (Fig. 1, middle). Thus,
two-photon excitation is a second way to obtain depth resolution and
suppression of out-of-focus fluorescence. Different from one-photon excitation
with confocal detection, which avoids out-of focus detection, two-photon
excitation avoids out-of-focus excitation.
Third, the fact that there is no
substantial excitation outside the focal plane makes two-photon excitation save
to live cells and tissue. Photodamage can occur only in the focal plane, and
not, as in the case of one-photon excitation, in the entire irradiated sample
volume. Detrimental effects on the viability of life cells and tissue are
therefore smaller than for one-photon excitation.
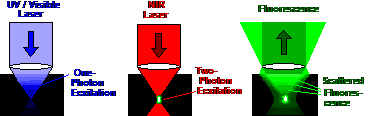
Fig. 1: Left: One-photon excitation. The effective excitation power
decreases rapidly with increasing depth. Middle: Two-photon excitation. The NIR
laser penetrates deeply into the sample. Right: The fluorescence from a deep
focus is scattered on the way out of the sample. It leaves the back aperture of
the microscope lens in a wide cone.
The fact that the fluorescence signal comes
only from the focus leads to a another advantage of two-photon excitation: A
two-photon microscope is able to detect photons which are scattered on the way
out of the sample. Scattered photons leave the back aperture of the microscope
lens in a wide cone of light (Fig. 1, right). In a one-photon system with a descanned
confocal beam path these photons would be lost. They can neither be fed through
the narrow beam path of the scanner, nor can they be focused into a pinhole. In
a two-photon microscope, however, there is no out-of focus excitation and,
consequently, no need to use a pinhole. Two-photon microscopes therefore divert
the fluorescence from the excitation beam directly behind the microscope lens
and project it on a large-area detector [3]. The principle is called
non-descanned (or direct) detection. A two-photon microscope with
non-descanned detection not only excites fluorescence in deep sample layers, it
also detects these photons efficiently.
Upgrading the DCS-120 Scanner for Multiphoton FLIM
Coupling the Ti:Sa Laser into the Scanner
Fibre coupling, as it is used for the
visible lasers of the DCS -120 system, cannot be used for the Ti:Sa laser
beam. The spectral dispersion in the glass would increase the pulse width so
much that efficient multiphoton excitation would not be obtained. The Ti:Sa
beam must therefore be free-beam coupled into the scanner. Fortunately, this
does not require changes in the scanner optics because the laser inputs are
designed for parallel beams [1]. Input 2, i.e. the input for the laser of
longer wavelength, must be used for the Ti:Sa laser, see Fig. 2.

Fig. 2: Input
of the Ti:Sa laser into the DCS‑120 scanner
Alignment of the laser beam with respect to
the scanner is critical, especially if also the confocal detectors are to be
used for multiphoton FLIM. It is therefore recommended to place the laser and
the microscope with the scanner on the same table. The scan head should be
attached to the table by a metal bracket. The laser beam should be fed via two
mirrors. One should be close to the laser, the other close to the scanner.
Adjustments at the first mirror then mainly influences the lateral beam
position, adjustment at the second mirror the beam angle.
Main Dichroic Beamsplitter of the Scanner
The main dichroic beamsplitter of the
scanner must be replaced with one that reflects the Ti:Sa Laser. A suitable beamsplitter
is available from bh. It reflects both the Ti:Sa laser and a 405 nm ps
diode laser, see Fig. 3.

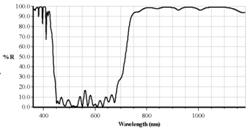
Fig. 3: Left:
Main dichroic beamsplitter block of DCS-120. Right: Characteristics of
multiphoton dichroic
Confocal Detectors
For reasons described above confocal
detection is not the best solution for multiphoton excitation. If the confocal
detectors are to be used for whatever reasons blocking filters for the Ti:Sa
laser wavelengths must be placed in the detection beam paths. The filters can
be screwed directly into the C-Mount adapters of the detectors, see Fig. 4.
Suitable filters and adapter rings are available from bh.

Fig. 4: Inserting
a laser blocking filter in front of the detector
Non-Descanned Detectors
Non-descanned detectors are attached via an
additional port of the microscope. The easiest solution is to use the
fluorescence-lamp port of the microscope. The principle is shown in Fig. 5. A
dichroic beamsplitter is inserted in the filter carousel of the microscope.
This beamsplitter lets the Ti:Sa laser pass but diverts the fluorescence to the
lamp port. The beamsplitter is switched into the beam path only for two-photon
excitation with non-descanned detection.
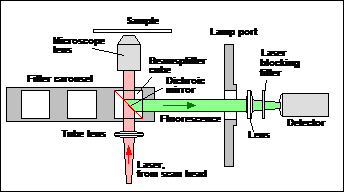
Fig. 5:
Principle of non-descanned detection optics (inverted microscope)
A lens in the non-descanned detection path
projects a de-magnified image of the microscope lens on the detector. The laser
wavelength is blocked by a filter in front of the detector, see Fig. 4. Please
note that this filter is absolutely required. The dichroic mirror in the
beamsplitter cube is not sufficient to reject the laser light.
In some cases, e.g. for the Zeiss Axiovert
and Axio Observer microscopes, the complete lamp port optics (with an
additional lens in front of the detector) can be used to project the
fluorescence signal on the detector. Adapters to the Zeiss lamp port are
available for all bh detectors.

Fig. 6: HPM‑100‑40
detector attached to lamp ort of Zeiss Axio Observer
Emission filters can be inserted directly
in front of the non-descanned detector. However, changing filters in this place
may be awkward. Filters should therefore be better placed in the filter sliders
some microscopes have in the path to the lamp port. If no such filter slider is
available we recommend to insert the emission filter in the output of the two-photon
beamsplitter cube in the microscope filter carousel. Several cubes, each with a
two-photon dichroic and a different filter, can be placed in the filter
carousel. If several non-descanned detectors are connected via a bh
beamsplitter assembly no emission filter in the microscope is needed. In that
case, filter cubes with different beamsplitter-filter combinations are used in
the beamsplitter assembly.
Multiphoton FLIM Procedure
The general procedure of recording
multiphoton FLIM is the same as for one-photon FLIM [1]. Put the sample in the
microscope. Switch the microscope beam path to the eyepieces (see below, laser
safety considerations). Turn the filter carousel to move the two-photon
dichroic out of the beam path. Bring it into focus and move it in the desired
position. Then turn the multiphoton dichroic in and switch the microscope beam
path to the scanner. Start the Preview mode. Note that the focus for the Ti:Sa
laser wavelength may be not exactly the same as for visible light. Focus into
the desired sample plane, and zoom into the desired image area. Adjust the laser
power. Then stop the preview, and start a FLIM recording with the desired
number of pixels. Let the acquisition run until you have reached a satisfactory
signal-to-noise ratio.
Please note that a non-descanned detector
detects from a much larger area than a confocal detector. It is therefore
mandatory to keep daylight out of the microscope. Unfortunately, normal
microscopes are not designed to for low daylight leakage. Therefore, cover the back
of the sample with piece of black cardboard, cover the complete microscope with
black cloth, and turn off the room lights before you start the FLIM
acquisition.
Results
Multiphoton NDD FLIM was tested with a
standard DCS‑120 system attached to a Zeiss Axio Observer Z1. The NDD
detector was attached to the lamp port of the Axio Observer as shown in Fig. 6
The scanner was modified with the dichroic beamsplitter shown in Fig. 3. The
TCSPC electronics was the standard SPC‑152 package of a bh Simple-Tau 152
system [4]. The NDD detector was a HPM‑100‑40 hybrid detector [4, 5].
Fig. 7 shows a two-photon NDD FLIM image of
the Convallaria sample that is routinely delivered with the DCS‑120
system. The convallaria is easy to use. It is easily visible in transmitted
light through the eyepieces and excites brightly at any laser wavelength from
780 nm to about 900 nm. No more than a few percent of the laser power
is needed to obtain a count rate on the order of 1 MHz. A typical result
is shown in Fig. 7.

Fig. 7: Two-photon FLIM of a convallaria sample, mean lifetime of
double-exponential decay model. 256x256 pixels, 256 time channels. Excitation
800 nm. Zeiss Axio Observer, x20 NA=0.6 lens, detection through lamp port,
HPM-100-40 detector.
To demonstrate the capabilities of
multiphoton NDD FLIM for realistic samples we used unstained samples of fresh
pig skin. Skin is highly scattering. When looking at the sample through the eyepieces
almost nothing can be seen. You may even have problems to find the surface of
the sample. Nevertheless, the two-photon exited autofluorescence FLIM images
are surprisingly rich in detail. Results for different depth in the sample are
shown in Fig. 8. bh SPCImage data analysis software [1, 2] with a
triple-exponential model was used to fit the decay data. The lifetimes shown
are amplitude-weighted averages of the decay components. Each image was
acquired at an average count rate of 1 MHz. The acquisition time was about
two minutes per image. No decrease in the count rate was observed over this
time, i.e. no photobleaching occurred.
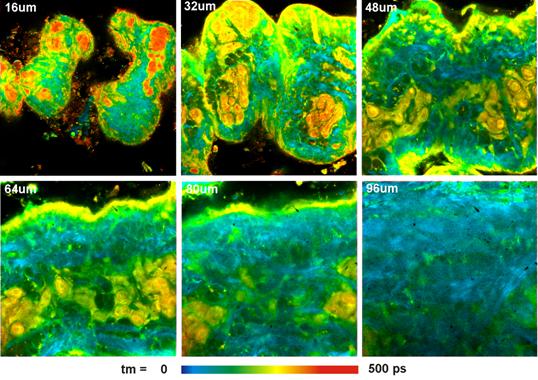
Fig. 8: Pig skin, autofluorescence, image in different depth in the
sample. Amplitude-weighted lifetime of triple-exponential decay model.
Excitation 805 nm, 512x512 pixels, 256 time channels. Zeiss Axio Observer
Z1, Water C apochromate NA=1.2, detection through lamp port, HPM‑100-40
hybrid detector.
The quality of the data shown in Fig. 8 is
so good that colour-coded images of individual parameters of the
triple-exponential can be generated. Of particular interest are the
contribution of the ultra-fast decay component, q1, and the ratio of the
amplitudes of the medium and slow component, a1/a2. The fast component is
dominated by SHG. SHG is generated by collagen and elastin. q1 therefore shows
the amount of these tissue constituents. The slow and medium decay component
are dominated by bound and unbound NADH. a1/a2 therefore gives insight into
NADH binding. The corresponding images are shown in Fig. 9.
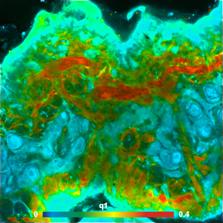
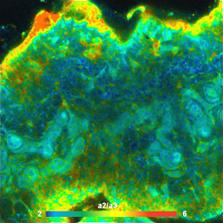
Fig. 9: Images of individual decay parameters of a triple-exponential fit
to the data. Left: Relative intensity contribution, q1, of the fastest decay
component. Right: Amplitude ratio, a2/a3, of the medium and slow decay component.
Depth 48 um below the surface.
Laser Safety Considerations
When using the DCS‑120 for
multiphoton excitation, please obey to the usual laser safety rules. In
particular, make sure that the laser light cannot be reflected back to the
eyepieces. That means in practice that microscopes with 50/50 beamsplitters
between the eyepiece and the scanner port should not be used, or the 50/50
position be blocked. Moreover, make sure that laser light is not reflected from
optical surfaces into the environment. In particular, light reflected off the
laser intensity regulators of the DCS‑120 scan head should be blocked by
a suitable beam stop. Moreover, we suggest to attenuate the laser power down to
about 20% directly behind the laser output by a suitable beamsplitter. 20% are
less dangerous than the full laser power, but are more than enough to obtain
FLIM from any reasonable sample. The beam splitter has the side effect that the
remaining 80% of the laser beam can be used for other applications without the
need of switching any optical elements in the beam.
Concluding Remarks
The DCS-120 system can be upgraded with a
Titanium-Sapphire laser and non-descanned detectors to obtain a multiphoton NDD
FLIM system. The upgraded system records autofluorescence images from tissue
layers as deep as 100 um within mammalian skin. The sensitivity of the
system is sufficient to obtain a count rate of 1 MHz from unstained pig
skin. No drop in the count rate was observed within a time of several minutes.
This indicates that no photobleaching occurred at the excitation power used.
The pig skin images shown above were
recorded within an acquisition time of two minutes. The long acquisition time
was used to obtain high-quality data suitable for triple-exponential decay
analysis. Please note also that the image format used was 512x512 pixels. For
lower pixel numbers correspondingly shorter acquisition time is sufficient to
obtain the same lifetime accuracy. For 256x256 pixels similar accuracy can be
obtained in 30 seconds, for 128x128 pixels in about 8 seconds.
Please note that the efficiency of a
two-photon microscope significantly depends on the microscope lens. Lenses of
high NA should generally be preferred. High NA not only results in a smaller
excited volume and thus in higher excitation efficiency, it also collects a
larger fraction of the fluorescence. High NA also avoids distortion of the
fluorescence decay functions by rotational depolarisation [4].
Please note that refractive-index mismatch
has a strong influence on the excitation efficiency in deep sample layers. The
excited volume remains small only if refractive-index mismatch is avoided. The
refractive index of biological tissue or cells is closer to water than to
immersion oil. Therefore, water immersion lenses usually give best results on
live samples, oil immersion lenses are better for fixed samples.
References
1. Becker & Hickl GmbH, DCS-120 Confocal Scanning FLIM Systems,
user handbook. www.becker-hickl.com
2.
Becker & Hickl GmbH, SPCImage Data
Analysis Software for Fluorescence Lifetime Imaging Microscopy. User handbook. Available on www.becker-hickl.com
3. Becker & Hickl GmbH, Non-Descanned FLIM Detection in
Multiphoton Microscopes. Application note, available on www.becker-hickl.com
4. W. Becker, The bh TCSPC Handbook, 4th edition (2010), available on
www.becker-hickl.com
5. Becker, W., Su, B., Weisshart, K. & Holub, O. (2011) FLIM and
FCS Detection in Laser-Scanning Microscopes: Increased Efficiency by GaAsP
Hybrid Detectors. Micr. Res. Tech. 74, 804-811
6.
W. Denk, J.H. Strickler, W.W.W. Webb, Two-photon laser scanning fluorescence microscopy, Science 248,
73-76 (1990)
7. M.
Göppert-Mayer, Über Elementarakte mit zwei Quantensprüngen, Ann. Phys. 9, 273-294 (1931)
8. T. Wilson, C. Sheppard, Theory and practice of scanning optical
microscopy. Academic Press (1984)
