Label-Free Multiphoton FLIM of Moving Bacteria
Wolfgang
Becker, Axel Bergmann, Julius Heitz, Becker & Hickl GmbH, Berlin, Germany
Abstract: Live bacteria are often difficult to
image in their natural environment because they are extremely mobile. To obtain
reasonable image quality extremely short image acquisition time has to be used
which, however, leads to pixel photon numbers insufficient for accurate
lifetime analysis. Higher excitation power does not solve the problem because
it impairs the viability of the bacteria or even causes severe photodamage. We
therefore developed a technique that records a sequence of fast single-frame
images, identifies the bacteria in these images by phasor analysis, and
combines the photons of the corresponding pixels in a single decay curve of
high photon number. This curve is processed by multi-exponential decay analysis
in the time-domain, yielding precision multi-exponential decay parameters of
the bacteria.
Autofluorescence FLIM
Biological material contains a number of fluorescent
compounds which can be used for fluorescence imaging. The most interesting one
is NAD(P)H (nicotinamide adenine (pyridine) dinucleotide). NAD(P)H is not only
involved in the metabolism of the cell, its fluorescence also depends on the
type of the metabolism. The effect is difficult to see by spectral imaging, but
can reliably be resolved by FLIM. It then reveals itself by different amounts
of fast and slow decaying fluorescence [1, 13, 18]. NAD(P)H FLIM - or NAD(P)H FLIM combined
with FAD FLIM - is therefore also called 'Metabolic FLIM' [5, 6]. It is being
used in a large number of life-science applications [11, 12, 15, 17, 19]. Please see [1] for an
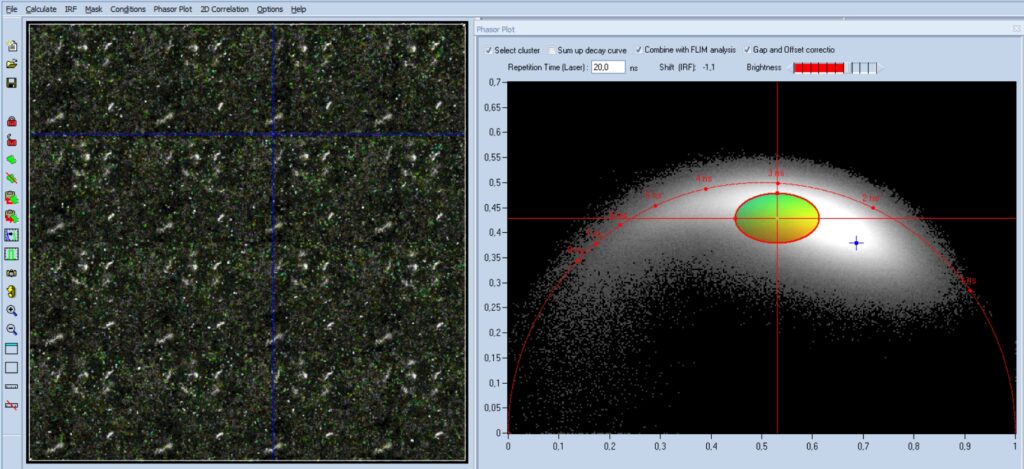
overview on the literature. A FLIM image of Streptococcus Salivarius
bacteria is shown in Fig. 1. The decay curves show the typical multi-exponential
behaviour of NAD(P)H. The fast component has a decay time of 361 ps and
comes from free NAD(P)H. The slow fluorescence is split in two components of 1.956 ns
and 5.395 ns and comes from bound NADPH and bound NADH [10].
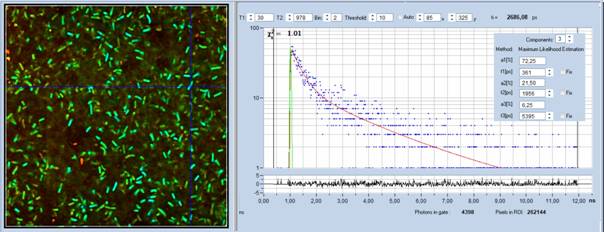
Fig. 1: NADH FLIM image of Streptococcus Salivarius. bh DCS-120 MP
FLIM system, excitation at 785 nm, detection from 440 nm to 480 nm.
Decay curve and decay parameters at cursor position shown in the right.
NAD(P)H imaging requires an excitation
wavelength of 340 to 380 nm. Wavelengths this short are difficult to
handle in a microscope. The transmission of the optics is far from ideal, and
there are large aberrations which impair the spatial resolution. NAD(P)H FLIM
is therefore usually performed by two photon excitation. Moreover, metabolic
FLIM has to be performed on live cells and tissues with intact metabolism. The
applicable excitation power is therefore very limited, and the fluorescence
intensities are low. In principle, this is no problem for bh's TCSPC FLIM [1, 2]. The timing stability of the technique
is so high that technically the acquisition time is virtually unlimited. A
sufficient number of photons can therefore always be acquired by simply
extending the acquisition time. Typical acquisition times are in the range from
10 seconds for medium lifetime accuracy and low pixel numbers to several
minutes for precision measurements. This is not very long but can cause a
problem if the objects of interest in the sample are moving.
The Problem
Motion in live samples is the nightmare of
any FLIM user. To acquire enough photons for precision decay analysis a large
number of frames have to be accumulated. This is no problem as long as the
objects in the sample stay in place for the entire acquisition time. However, if
the objects under investigation are moving, as bacteria do, the image is
smeared out, and the decay data of the bacteria are mixed with signals from the
substrate. The first idea in these cases is usual to pull up the excitation
power and thus increase the photon flux. A given accuracy of the decay data
would then be reached in a shorter acquisition time. Unfortunately, the options
of this approach are very limited. High excitation power quickly causes invasive
effects or even catastrophic damage in the sample, rendering the FLIM results
useless. This is especially the case in autofluorescence experiments where the
emission from the cells is weak.
An example is shown in Fig. 2. The images
show live Escherichia coli bacteria on the substrate they were cultured
on. The data were recorded with a bh DCS-120 MP Fibre-Laser system at 785
nm excitation wavelength. The bacteria are extremely mobile. Recorded at an
acquisition time of 1.5 seconds the bacteria are still marginally visible
(left image). However, the photon numbers in the pixels are not sufficient for
multi-exponential decay analysis. Another problem that shows up in Fig. 2 is
background fluorescence from the substrate. For immobile object it is
suppressed by the depth resolution of the two-photon excitation process. For
objects that move during the acquisition, however, fluorescence of the
background is mixed into the fluorescence of the objects.
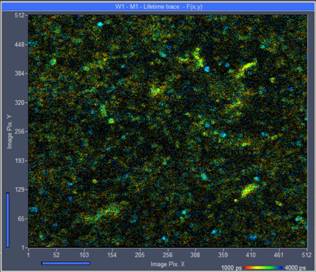
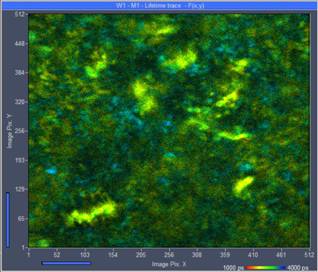
Fig. 2: Left: Escherichia coli bacteria culture, single scan of 1.5 second
duration. The bacteria are marginally visible, but there are not enough photons
for accurate lifetime analysis. Right: Same sample, acquisition time 24 s.
There are enough photons per pixel but motion has rendered the bacteria
invisible.
With the acquisition time increased to 24
seconds (Fig. 2, right) the motion has totally smeared out the image detail.
Only immobile bacteria are still visible. These bacteria have unusually long
lifetimes. They are probably not the viable ones and are thus not interesting
for further investigation.
As mentioned above, it is usually attempted
to solve the problem by higher excitation power. Higher excitation power
yields, of course, a higher rate of fluorescence photons and thus a better image
in a shorter period of time. But this comes at a price. Fig. 3 shows a
recording with 2.5 times increased excitation power. This leads to a 6.52 fold
increase in photon rate. No doubt bacteria are better visible than in Fig. 2,
but a large number of spots with extremely short lifetimes (red in the image)
have appeared. These are burn marks which indicate that severe photodamage has
occurred. Of course, there are still bacteria which appear to be intact.
However, with severe photodamage present nearby, the decay data and,
especially, the metabolic information derived from them cannot be trusted.
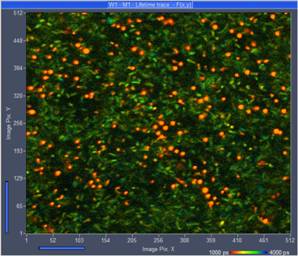
Fig. 3: FLIM recording at 2.5 x increased laser power. The red spots
of ultra-short lifetime indicate sample damage. Online-lifetime display of
SPCM, red to blue = 1 ns to 4 ns.
The problems shown in Fig. 2 and Fig. 3
have led to an avalanche of demands for faster FLIM techniques. These solutions
are designed to work at high photon flux, but at the expense of photon
efficiency [4, 14]. Of course, the approach was not successful, leading to the
conclusion that FLIM is not a suitable technique for molecular imaging. Of
course, this is wrong. Where is the mistake?
The limitation of acquisition speed is in
the ability of the sample to deliver high photon rates, not in the counting
capability of the FLIM technique. Increasing the counting capability at the
expense of photon efficiency makes the situation worse. More photons are needed
to reach a given accuracy level, with the result that damage effects become
even more severe. Unless the samples are extremely bright and stable (which
is not the case in molecular imaging applications [1]) TCSPC FLIM is and
remains to be the fastest technique because it needs less photons than 'fast'
FLIM techniques.
The Solution
Image Segmentation by Phasor Plot
So, how can the problem of motion in the
samples be solved? Obviously, not with a faster FLIM technique. A possible way
to obtain decay information from the moving bacteria would be to record a
single, fast scan, identify the bacteria in the image, and sum up the decay
data of all pixels which are located inside the bacteria. The resulting curve
will contain a significantly higher number of photons than a single pixel.
How can the bacteria be identified in the
image of a single scan? A simple (and readily available) way is the phasor plot
of bh's SPCImage NG FLIM data analysis, please see [1, 7, 9]. The phasor plot
reacts very sensitively to differences in the signature of the decay data in the
pixels. Pixels of similar decay signature are located close to each other in
the phasor plot. Even if the photon number is too low for pixel-by-pixel
analysis the phasor range of the bacteria can be indentified. The phasor range of
interest is then marked by the operator, and the corresponding pixel are
back-annotated in the image. The decay data in these pixels are summed up, and
the resulting curve is analysed by the normal time-domain fit routine.
The procedure is illustrated in Fig. 4. A
single-frame image is shown on the left. As expected, standard decay analysis does
not deliver precision decay data of the bacteria, see decay data lower right.
Nevertheless, the maximum-likelihood fit of SPCImage NG delivers an approximate
lifetime (shown by colour) in the image. The phasor plot (upper right) shows a
cloud of phasor values for the individual pixels of the scan. Selecting a range
of phasors of the same colour as the bacteria (compare FLIM image on the left)
selects the pixels inside the bacteria.
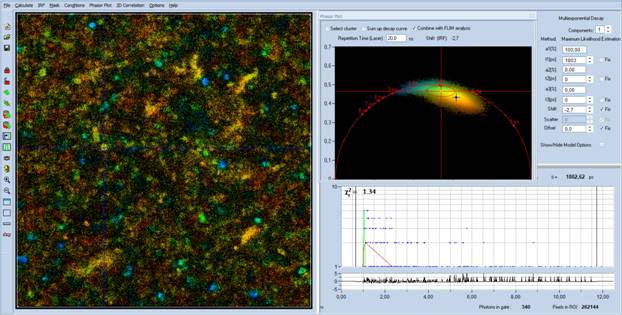
Fig. 4: Single-frame data loaded into SPCImage NG. Left: Lifetime image,
approximate lifetime. Upper right: Phasor plot. Lower right: decay curve at
cursor position.
When an appropriate phasor range has been
selected, the 'Select Cluster' function is activated, see Fig. 5, upper left of
phasor plot. This suppresses analysis of pixels outside the selected phasor
range. The result can be seen in the lifetime image. It indeed selects the bacteria,
showing that the phasor range was estimated correctly. Now activate 'Sum Up
Decay Curve'. This combines the decay data of all pixels of the selected phasor
range into a single decay curve. The resulting curve has a large number of
photons (see lower right) and can be analysed at high accuracy. In Fig. 5 a fit
with a double-exponential model was used. It delivers the average decay
parameters of the bacteria, see upper right. The component amplitudes, a1 and
a2, and the component lifetimes, t1 and t2, are shown in the upper right. All
parameters are well in the range expected for NAD(P)H.
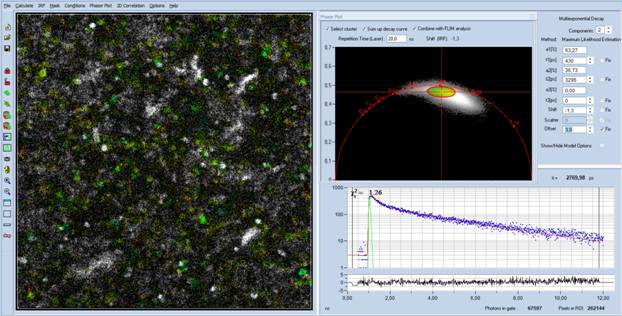
Fig. 5: Data from Fig. 4, 'Select Cluster' and 'Sum Up Decay Curve'
active. The decay data of the selected phasor range are combined into a single
decay curve, representing the average decay curve of the bacteria. The decay
parameters (upper right) are as expected for NAD(P)H.
Mosaic FLIM with Image Segmentation by Phasor Plot
The method described above works well if
there is a sufficient number of objects in the image and if the expectations to
the decay accuracy are moderate. If this is not the case a more powerful
procedure can be used. The idea behind it is: If image segmentation in a single
scan is effective to derive a high accuracy decay curve then segmentation in
several scans will be even more effective.
We have shown above that a single fast scan
delivers an reasonably sharp image in which moving objects can be identified. A
second fast scan will deliver a sharp image as well. However, the bacteria will
have moved to different places, some of them may have moved out of the focal
plane, and other may have moved in. In other words, every scan shows a
different image. If the objects are identified in all these single-scan images
and the decay data of the corresponding pixel be summed up the increase in
photon number will be even more dramatic than in a single scan. Moreover, in
contrast to the approach described above the Mosaic approach works also for a
single object in the field of view which is either moving or changing its shape
[8].
The procedure is illustrated in Fig. 6 to Fig.
8. A mosaic of FLIM images is recorded by the Mosaic recording function of SPCM
data acquisition software [1].
The result is shown in Fig. 6. Please note that it is not required to move the
sample from one mosaic element to the other - the images of subsequent scans
are different anyway. The entire mosaic is loaded into SPCImage NG, a
preliminary FLIM analysis is performed, and the phasor plot is opened, see Fig.
7. The phasor range formed by the bacteria is selected and the photons of the
corresponding pixels are summed up, see Fig. 8. The result is a decay curve
that has an accuracy previously known only for cuvette-based measurement. A fit
of the resulting decay curve with a triple-exponential model delivers the decay
parameters of the bacteria, see Fig. 8, upper right. The fit even splits the
slow fluorescence into two decay components, known to come from bound NADH and bound
NADPH [10].
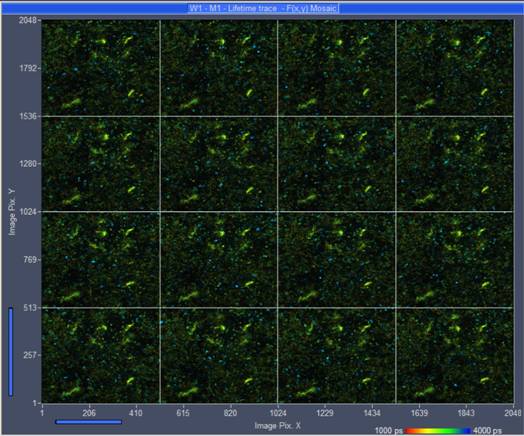
Fig. 6: 4x4
Mosaic of single scans, recorded by Mosaic Imaging function of SPCM data acquisition
software. Displayed by online-lifetime function of SPCM. The data structure of
mosaic resembles that of a single, large FLIM image.
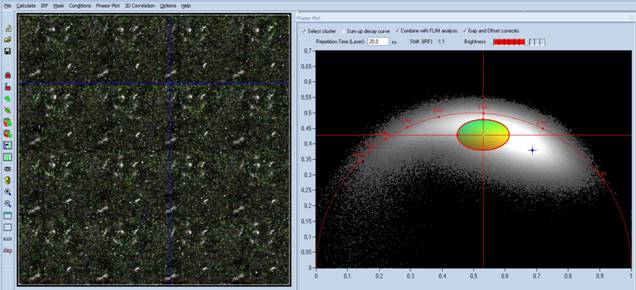
Fig. 7: 4x4
mosaic, loaded into SPCImage NG. Phasor plot enabled, phasor range of bacteria
selected, selected pixels back-annotated in the FLIM image.
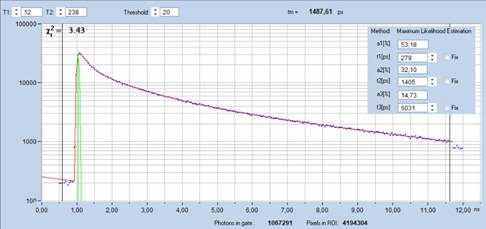
Fig. 8: Decay
curve obtained by summing up decay data of pixels selected by phasor plot. Fit
with SPCImage NG, triple-exponential 'incomplete-decay' model. Total number of
photons 1,067,291, Decay parameters shown upper right.
Summary
Molecular imaging by FLIM often faces the
problem that the objects of interest in the sample are moving. Reasonable
images are then only achieved by imaging with short acquisition time. However,
molecular imaging not only has to be performed on live samples but is also
connected to low fluorophore concentrations and fluorophores of low quantum
yield. The number of photons obtained from the sample within a short period of
time is then not sufficient for accurate multi-exponential decay analysis. A
solution of the problem lies in a combination of time-domain analysis and
phasor analysis. Pixels within the objects are identified via the phasor plot
and the decay data of these pixels are combined into a single curve. The
combined data contain enough photons for decay analysis. An even more dramatic
improvement is obtained by combining this procedure with the Mosaic Imaging
function of the bh FLIM systems. The photon number obtained this way is
comparable with the photon number in cuvette-based fluorescence-lifetime
measurements. Consequently, precision analysis of the decay curve is possible.
We show that the data quality obtained this way is so high that precision
triple-exponential decay analysis of NAD(P)H is possible.
References
1.
W. Becker, The bh TCSPC handbook. Becker &
Hickl GmbH, 9th ed. (2021). pdf available on www.becker-hickl.com. Please contact bh for printed copies.
2.
W. Becker, The bh TCSPC Technique. Principles
and Applications. Becker & Hickl GmbH, pdf available on
www.becker-hickl.com.
3.
W. Becker, DCS-120 Confocal and Multiphoton FLIM
Systems, user handbook, 9th ed., Becker & Hickl GmbH (2021). Available on
www.becker-hickl.com
4.
W. Becker, S. Smietana, Fast-Acquisition TCSPC
FLIM: What are the Options? Application note, available from
www.becker-hickl.com
5.
W. Becker, A. Bergmann, L. Braun, Metabolic
Imaging with the DCS-120 Confocal FLIM System: Simultaneous FLIM of NAD(P)H and
FAD, Application note, available on www.becker-hickl.com (2018)
6.
W. Becker, R. Suarez-Ibarrola, A. Miernik, L.
Braun, Metabolic Imaging by Simultaneous FLIM of NAD(P)H and FAD. Current
Directions in Biomedical Engineering 5(1), 1-3 (2019)
7.
Becker & Hickl GmbH, SPCImage next
generation FLIM data analysis software. Overview
brochure, available on www.becker-hickl.com
8.
Precision Fluorescence-Lifetime Imaging of a
Moving Object. Application note, available on www.becker-hickl.com.
9.
Becker & Hickl GmbH, New SPCImage Version
Combines Time-Domain Analysis with Phasor Plot. Application note, available on
www.becker-hickl.com
10.
T. S. Blacker, Z. F. Mann, J. E. Gale, M.
Ziegler, A. J. Bain, G. Szabadkai, M. R. Duchen, Separating NADH and NADPH
fluorescence in live cells and tissues using FLIM. Nature
Communications 5, 3936-1 to -6 (2014)
11.
A. A. Gillette, C. P. Babiarz, A. R. Van
Dommelen, C. A. Pasch, L. Clipson, K. A. Matkowskyj, D. A. Deming, M. C. Skala,
Autofluorescence Imaging of Treatment Response in Neuroendocrine Tumor
Organoids. Cancers (Basel). 13(8), 1873, 1-17 (2021)
12.
S. Kalinina, V. Shcheslavskiy, W. Becker, J.
Breymayer, P. Schäfer, A. Rück, Correlative NAD(P)H-FLIM and oxygen
sensing-PLIM for metabolic mapping. J. Biophotonics 9(8):800-811 (2016)
13. J.R. Lakowicz, H. Szmacinski, K. Nowaczyk, M.L. Johnson,
Fluorescence lifetime imaging of free and protein-bound NADH, PNAS 89,
1271-1275 (1992)
14.
X. Liu, D. Lin, W. Becker, J. Niu, B. Yu, L.
Liu, J. Qu, Fast fluorescence lifetime imaging techniques: A review on
challenge and development. Journal of Innovative Optical Health
Sciences, 1930003-1 to -27 (2019)
15.
M. M. Lukina, V. V. Dudenkova, N. I. Ignatovaa,
I. N. Druzhkova, L. E. Shimolina, E. V. Zagaynovaa, M. V. Shirmanova, Metabolic
cofactors NAD(P)H and FAD as potential indicators of cancer cell response to
chemotherapy with paclitaxel. BBA General Subjects 1862, 1693-1700
(2018)
16.
M. M. Lukina, L. E. Shimolina, N. M. Kiselev, V.
E. Zagainov, D. V. Komarov, E. V. Zagaynova, M. V. Shirmanova, Interrogation of
tumor metabolism in tissue samples ex vivo using fluorescence lifetime imaging
of NAD(P)H. Methods Appl. Fluoresc. 8, 014002, 1-11 (2020)
17.
M.N. Pastore, H. Studier, C.S. Bonder, M.S.
Roberts, Non-invasive metabolic imaging of melanoma progression. Exp.
Dermatol. 26, 607614 (2017)
18. R.J. Paul, H. Schneckenburger, Oxygen concentration and the
oxidation-reduction state of yeast: Determination of free/bound NADH and
flavins by time-resolved spectroscopy, Naturwissenschaften 83, 32-35
(1996)
19.
A.T. Shah, K.E. Diggins, A.J. Walsh, J.M. Irish,
M.C. Skala, In vivo autofluorescence imaging of tumor heterogeneity in response
to treatment. Neoplasia 17, 862-870 (2015)
Contact:
Wolfgang Becker
Becker & Hickl GmbH
Berlin, Germany
Nunsdorfer Ring 6-9
Email: becker@becker-hickl.com