Microsecond Decay FLIM: Combined Fluorescence and Phosphorescence Lifetime Imaging
Wolfgang Becker, Stefan
Smietana, Axel Bergmann, Becker & Hickl GmbH
Abstract. We present a lifetime imaging technique
that simultaneously records fluorescence and phosphorescence lifetime images
in laser scanning systems. It is based on modulating a high-frequency pulsed
laser synchronously with the pixel clock of the scanner, and recording the
fluorescence and phosphorescence signals by multi-dimensional TCSPC. Fluorescence
is recorded during the on-phase of the laser, phosphorescence during the
off-phase. The technique does not require a reduction of the laser pulse
repetition rate by a pulse picker, and eliminates the need of using excessively
high pulse power for phosphorescence excitation. Laser modulation is achieved
either by electrically modulating picosecond diode lasers, or be controlling
the lasers via the AOM of a standard confocal or multiphoton laser scanning
microscope.
Motivation of Using Phosphorescence Decay Imaging
There is a number of radiative relaxation
mechanisms which occur on a much longer time scale than fluorescence. The
commonly known one is phosphorescence, i.e. emission from the triplet state of
organic dyes. Phosphorescence of organic dyes is usually weak at room temperature
but can be strong at low temperatures, or if the dyes are embedded in a solid
matrix. Strong emission in the microsecond and millisecond range is obtained
also for lanthanide complexes [5] and organic complexes of ruthenium [7],
platinum [8], and palladium [8]. Of special interest for live-cell imaging is
that the luminescence of some of these complexes is strongly quenched by
oxygen. The dyes make then excellent oxygen sensors [6, 7, 8, 9, 10]. There are also possible applications as
FRET donors [5]. Moreover, slow emission is obtained from a large number of
inorganic compounds, quantum dots, nanoparticles, and semiconductors.
Technical Problems of Slow-Decay Imaging
The measurement of long-lifetime
luminescence by laser scanning systems faces a number of problems. The first one
is related to the excitation of the luminophore. Obviously, the laser pulse period
must be longer than the luminescence lifetime. The lifetime for ruthenium is in
the lower microsecond range; for europium and terbium dyes it can be in the
millisecond range. FLIM with these dyes requires a laser repetition rate no
faster than 100 kHz or 100 Hz, respectively. Generating such low
repetition rates with pulsed lasers of a laser scanning microscope can be a
problem. More important, reduction in repetition rate, for a given pulse power,
leads also to a reduction in average excitation power. Attempts to compensate
for the drop in average power by higher pulse power are limited by the capabilities
of the laser, and by nonlinear effects or even ionisation in the sample.
Moreover, any sample that emits phosphorescence necessarily also emits
fluorescence. Because fluorescence is fast the peak power of fluorescence
becomes very high. This causes transient overload effects in the detectors, preventing
the detection of phosphorescence in the first microseconds after the laser
pulse. A better way to obtain higher average power is therefore to use longer
laser pulse width. Unfortunately, this is not easily possible for most of the
lasers. Moreover, long laser pulse width is incompatible with multiphoton excitation.
The second problem is related to scanning.
The time the scanner stays within the excited sample volume must be longer than
the luminescence lifetime. If the scanner runs off the excited volume within
the luminescence decay time photons in the tail of the decay function would be
lost, and the recorded decay profile be distorted. Reasonable recording, even
of pure intensity images, can thus only be obtained by very slow scanning.
An third problem is aliasing of the laser
repetition rate with the pixel frequency: If there are only a few excitation
pulses within the pixel time the number of excitation pulses in the pixels
varies systematically. This induces Moiré effects in the images. The problem
can be solved by synchronising the laser pulses and the pixel frequency, but
there is usually no provision for this in a normal laser scanning microscope.
Without synchronisation, the pixel time must be at least 100 times longer than
the laser period. This leads to unacceptably long frame times.
Of course, the scanner problems can be
avoided by using wide-field excitation and detection with a gated camera.
However, abandoning scanning also abandons optical sectioning and depth
resolution. For most biological applications this is not acceptable.
Modulated Pulsed Laser Operation with TCSPC Recording
The problems described above are avoided by
the excitation principle shown in Fig. 1. A high-frequency pulsed laser is
used. However, the laser is not run continuously. Instead, it is turned on only
for a short period of time, ton, at the beginning of each pixel [2, 9]. For the rest of the pixel time the laser
is turned off. Within the on-time, ton, the laser excites
fluorescence, and builds up phosphorescence. Within the rest of the pixel dwell
time, toff, pure phosphorescence is obtained.
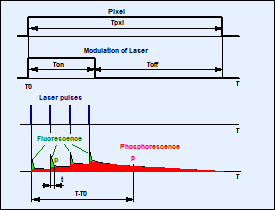
Fig. 1:
Principle of Microsecond FLIM
The modulation of the laser is controlled
by the bh FLIM system. In the DCS-120 confocal scanning system the laser
modulation signal is generated by using the laser-multiplexing features of the bh
GVD‑100 scan controller. The BDL‑SMC diode lasers of the DCS‑120
are electronically modulated by applying this signal to their /laseroff inputs
[3].
For other microscopes a bh DDG-210 card is
added to the FLIM system. The card is triggered by the pixel clock of the
microscope and generates the laser modulation signal. Modulation is obtained by
combining this signal with the beam blanking signal of the microscope. Laser
modulation is then obtained via the acousto-optical modulator of the
microscope. The modulation also acts on the Ti:Sa laser of a multiphoton microscope.
Thus, microsecond decay imaging becomes applicable also to deep-tissue imaging
by two-photon excitation and non-descanned detection.
Lifetime images are built up by using the
double-kinetic features of the SPC modules [1, 2]. The principle is shown in Fig.
2. For each photon, the SPC module determines the time, t, within the laser
pulse period, and the time, T-T0, after the modulation pulse. A
fluorescence lifetime image is obtained by building up a photon distribution
over the micro times, t, of the photons, and the scanner position, x,y,
during the Ton periods. The phosphorescence lifetime image is
obtained by building up a distribution over the time differences, T-T0,
between the photon times, T, and the laser on pulse edges, T0. The
spatial coordinates come from the scanner position in the moment of the photon
detection. Thus, fluorescence and phosphorescence lifetime images are obtained
simultaneously, in the same scan, and from photons excited by the same laser
pulses.
Multi-wavelength operation is possible by
using the routing (or multi-detector) capability of the bh SPC modules [1, 2].
In this case, photons are marked additionally with the wavelength channel in
which they were detected. Individual fluorescence and phosphorescence lifetime
images are then built up for the individual wavelength channels.
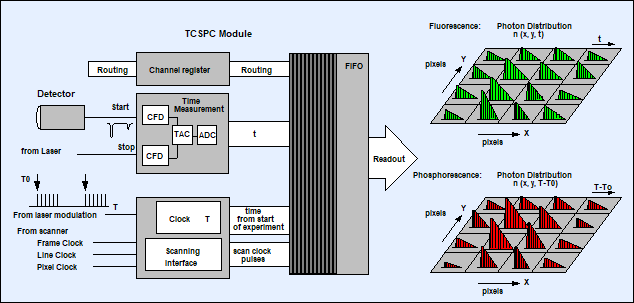
Fig. 2:
Simultaneous fluorescence and phosphorescence lifetime imaging. Photon times
are determined both with respect to the laser pulse period and with respect to
the laser modulation period.
Combined fluorescence / phosphorescence
decay imaging has been introduced with the version 9.0 release of the bh SPCM
software, April 2010. It is available for SPC‑150 modules and SPC‑830
modules manufactured later than May 20073, or serial numbers later than 3D0178.
TCSPC System Parameter Setup
Typical system setup parameters for the SPC
module are shown in Fig. 3. Phosphorescence imaging is obtained by using the
MCS (Multichannel Scaler) option in the FIFO Imaging mode. The MCS option
is selected in the Configure panel of the System Parameters, see Fig. 3, middle.
The timing reference for MCS imaging comes
from the laser modulation. A suitable pulse must be connected to a Marker
input at the 15-pin connector of the SPC module [2]. Three marker inputs are
available. Trigger defines which of the marker inputs is used as a timing
reference. Normally, markers 0, 1, and 2 are used for the pixel clock, line
clock, and frame clock from the scanner. The timing reference is then marker 3.
However, if the excitation pulses are synchronous with the pixels Trigger can
be identical with the pixel clock, i.e. Marker 0. Please make sure that the
marker input used for the trigger is enabled in the More Parameters panel of
the System Parameters, see Fig. 3 right.
The time channel width (Time per point) of
the MCS imaging mode can be any multiples of the macro time clock period. It is
defined by a number of Macro Time units. The number of points of the decay curves
within the pixels is defined by Points No.. The time range of the curves is
given by the product of Time per Point and Points No. It is displayed under
Time range. The recorded time interval can be shifted
by applying an Offset to the photon times. Both positive and negative offsets
are possible.
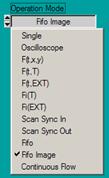
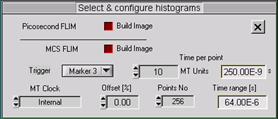
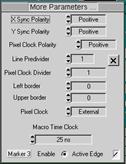
Fig. 3: Definition of MCS FLIM parameters. Left: Operation mode. Middle:
Definition of timing parameters and marker selection. Right: Scan format
parameters, macro time clock source, and marker enable.
Normally, MCS FLIM recording is performed
with the internal macro time clock of the SPC module, see Fig. 3, middle and
right. However, for special applications the SYNC frequency can be used. Using
the SYNC frequency does, of course, require that the SYNC signal has a constant
period, and is present also during the laser off intervals.
To obtain picosecond FLIM (either
separately or simultaneously with phosphorescence lifetime imaging) activate
the ps FLIM button in the configuration panel. The timing parameters for ps
FLIM are selected in the usual way via the TAC parameters of the SPC module [2].
Control of the Laser Modulation
Phosphorescence imaging requires that the
excitation laser is on/off modulated by a signal synchronous with the pixel
clock, as shown in Fig. 1.
In the Becker & Hickl DCS-120 confocal
scanning system [3]
pixel-synchronous laser modulation is obtained by using the existing laser-multiplexing
features of the scan controller. The DCS system has two ps diode lasers. For
phosphorescence lifetime imaging, one laser is used for excitation, the other
one is optically turned off. With pixel-synchronous laser multiplexing the
modulation scheme shown in Fig. 4, left is achieved.
The setup parameter panel for the DCS‑120
confocal scanner is shown in Fig. 4. Laser Multiplexing is set to Pixel,
and a turn-on time for the laser of 12.5% of the pixel time is set. In the
Scan Rate definitions the automatic scan rate selection is disabled, and a
pixel time a few times longer than the expected phosphorescence decay time is
used. Extremely long decay times may require an extension of the available
range of the scan rate. This can be obtained by defining a new Max Line Time
in the Scan Details panel, see Fig. 4, right.
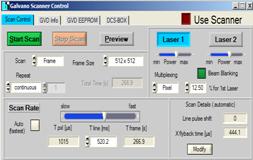

Fig. 4: DCS-120 system, scanner control parameters for phosphorescence
decay imaging. Left: definition of laser modulation and scan rate control.
Right: Scan Details, definition of maximum line time
For implementation of phosphorescence
lifetime imaging in other laser scanning microscopes the microscope must have a
pixel clock output from which the laser modulation signal can be derived.
Moreover, the microscope must allow for an input of the laser modulation signal
to its internal beam blanking. To generate the laser modulation signal we use a
Becker & Hickl DDG‑210 programmable pulse generator card. The DDG-210
is triggered by the pixel clock. It delivers a laser modulation signal of
programmable width, which is fed back into the beam blanking system of the
microscope, see Fig. 5. A second signal is generated to indicate to the TCSPC
module whether the laser is on or off. It is used by the TCSPC module to route
fluorescence and phosphorescence photons into separate memory blocks [2]. The
routing signal is slightly delayed with respect to the modulation signal to
account for the delay in the opto-acoustic modulator (AOM) of the microscope.
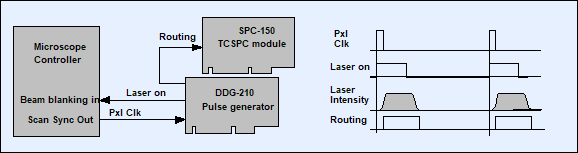
Fig. 5: On-off modulation of laser in other scanning systems than the DCS‑120.
The pixel clock of the microscope triggers the generation of a laser-on pulse
in the DDG-210 pulse generator module. The laser-on pulse controls the beam
blanking in the microscope. The AOM of the microscope responds to the beam
blanking with a few µs delay. A routing signal to the SPC-150 TCSPC module
indicates when the laser is on. Connection diagram shown left, pulse diagram
right.
Results
DCS-120 confocal FLIM system
An example of phosphorescence lifetime imaging
with the DCS‑120 FLIM system is shown in Fig. 6 and Fig. 7. The images
were obtained from particles of an inorganic dye. A BDL‑405 SMC
laser was used for excitation; the signals were recorded by the Simple-Tau 152
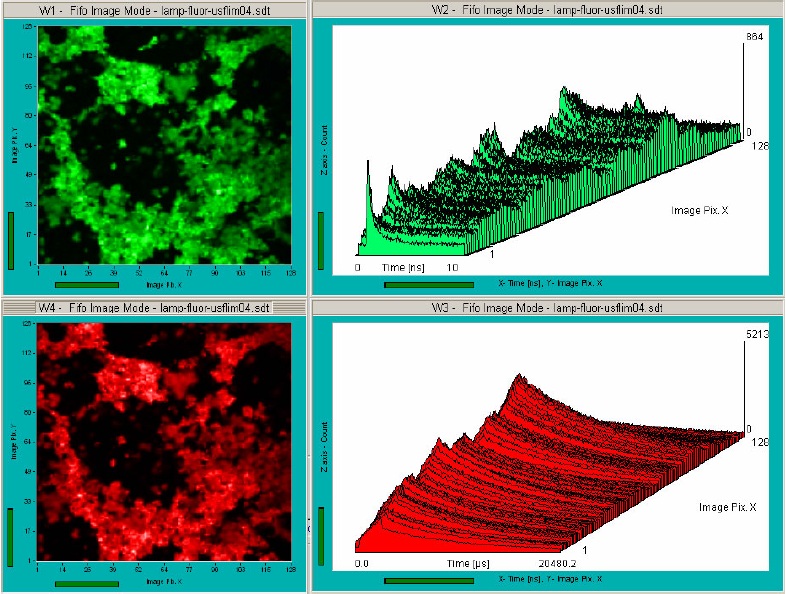
system of the DCS‑120. Fig. 6 shows images displayed by the SPCM data acquisition
software. Lifetime images analysed with the bh SPCImage FLIM analysis software
are shown in Fig. 7.

Fig. 6: Fluorescence image (upper left) and phosphorescence image (lower
left) recorded simultaneously. ps decay curves and µs decay curves over a
horizontal section of the image are shown upper right and a lower right,
respectively. Particles of an inorganic luminophore, BDL-405SMC laser,
Simple-Tau 152 FLIM system, SPCM data acquisition software.

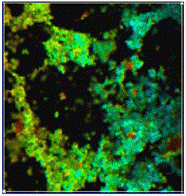
Fig. 7: Fluorescence lifetime image (left) and phosphorescence lifetime
image (right) obtained from the data shown above. Colour scale red to blue
500 ps to 4 ns and 10 to 15 ms, respectively. SPCImage data
analysis software.
Zeiss LSM 710 NLO Multiphoton Microscope
To demonstrate simultaneous recording of
fluorescence and phosphorescence by two-photon excitation we used a Zeiss
LSM 710 NLO multiphoton microscope. An excitation wavelength of
780 nm was used. The luminescence was collected through the non-descanned
beam path of the LSM 710. The photons were detected by a Becker & Hickl HPM‑100-40
hybrid detector and recorded by a Becker & Hickl SPC‑150 TCSPC FLIM
module [4]. Laser modulation was achieved as shown in Fig. 5.
As a sample we used yeast cells stained
with a ruthenium dye. The cells where kept in water in a cell dish, and a small
amount of tris(2,2-bipyridyl)dichlororuthenium(II)hexahydrate was added.
Intensity images obtained this way are
shown in Fig. 8. Depending on the display parameters set in the TCSPC software
different images can be displayed from the same data set recorded. The left
image shows the luminescence during the laser-on phases. It contains mainly
autofluorescence of the cells, with a small contribution of ruthenium
phosphorescence. The image in the middle shows the emission in the laser-off
phases. It contains only phosphorescence. Phosphorescence mainly comes from the
cell membrane to which the ruthenium dye binds. The image shown right shows the
sum of the fluorescence and phosphorescence intensity.

Fig. 8: Intensity images of yeast cells stained with a ruthenium dye.
Images from left to right: Fluorescence, phosphorescence, total emission. LSM
710 NLO, two-photon excitation at 780 nm, non-descanned detection,
HPM-100-40 hybrid detector, SPC-150 TCSPC FLIM module.
Lifetime images obtained from the same data
set are shown in Fig. 9. The fluorescence lifetime image is shown on the left,
the phosphorescence lifetime image on the right. Fig. 10 shows decay curves for
a selected spot in the images. The blue dots are the photon numbers in the
subsequent time channels, the red curve is a fit with a double-exponential
decay model, and the green curve is the effective instrument response function
(IRF). Please note that the IRF of the fluorescence decay is the laser pulse
convoluted with the detector response, whereas the IRF of the phosphorescence
decay is the waveform of the laser modulation.


Fig. 9: Fluorescence lifetime image (left) and phosphorescence lifetime
image (right) of yeast cells stained with
tris(2,2-bipyridyl)dichlororuthenium(II)hexahydrate. Amplitude-weighted mean
lifetime of double exponential fit to decay data. Same data set as in Fig. 8,
data analysis by Becker & Hickl SPCImage.
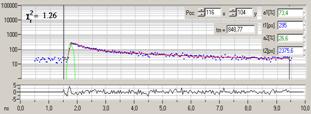
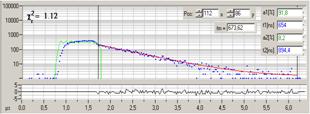
Fig. 10: Decay curves in selected spot of Fig. 9. Left: Fluorescence.
Right: Phosphorescence. Blue dots photon numbers in the time channels, red
curve fit with double-exponential decay model, green curve effective instrument
response function. Inserts: Amplitude-weighted lifetime and double-exponential decay
parameters. Decay parameters in ps for fluorescence and in ns for
phosphorescence.
Summary
The technique described above simultaneously
records fluorescence and phosphorescence lifetime images in confocal and
multiphoton laser scanning systems. It eliminates the requirement of using a
pulse picker for reduction of the pulse repetition rate, and avoids excessively
high pulse power at low excitation rate. The technique can directly be used in
the bh DCS‑120 confocal scanning FLIM system. It can easily be
implemented in other laser scanning microscopes if these give access to their
internal beam blanking control.
Potential applications are oxygen
concentration measurements with simultaneous monitoring of cell metabolism via
autofluorescence signals, identification of nanoparticles of sunscreens and
cosmetical products in the skin, and observation of possible migration of these
particles into deep skin layers or inner organs.
References
1. W. Becker, Advanced time-correlated single-photon counting techniques. Springer,
Berlin, Heidelberg, New York, 2005
2.
W. Becker, The bh TCSPC handbook. 3rd edition,
Becker & Hickl GmbH (2008), available on www.becker-hickl.com
3. Becker & Hickl GmbH, DCS-120 Confocal Scanning FLIM Systems,
user handbook. www.becker-hickl.com
4. Becker & Hickl GmbH, Modular FLIM systems for Zeiss
LSM 510 and LSM 710 laser scanning microscopes. User handbook,
available on www.becker-hickl.com
5. L.J. Charbonniere, N. Hildebrandt, Lanthanide complexes and quantum
dots: A bright wedding for resonance energy transfer. Eur. J. Inorg. Chem.
2008, 3241-3251 (2008)
6. H.C. Gerritsen, R. Sanders, A. Draaijer, Y.K. Levine, Fluorescence
lifetime imaging of oxygen in cells, J. Fluoresc. 7, 11-16 (1997)
7.
J.R. Lakowicz, Principles of Fluorescence
Spectroscopy, 3rd edn., Springer (2006)
8. A. Y. Lebedev, A. V. Cheprakov, S.
Sakadzic, D. A. Boas, D. F. Wilson, Sergei A. Vinogradov, Dendritic
Phosphorescent Probes for Oxygen Imaging in Biological Systems. Applied
Materials & Interfaces 1, 1292-1304 (2009)
9. S. Sakadic, E. Roussakis, M. A.
Yaseen, E. T. Mandeville, V. J. Srinivasan1, K. Arai, S. Ruvinskaya, A. Devor,
E. H. Lo, S. A. Vinogradov, D. A. Boas, Two-photon high-resolution measurement
of partial pressure of oxygen in cerebral vasculature and tissue. Nature
Methods 7(9) 755-759
10. M. Shibata, S. Ichioka, J. Ando, A. Kamiya, Microvascular and
interstitial PO2 measurement in rat skeletal muscle by phosphorescence
quenching. J. Appl. Physiol. 91, 321-327 (2001)